
Redalyc.Clonaje y expresión heteróloga de un fragmento de ... · El inserto escindido de 2,1 kb,...
12
Revista CENIC. Ciencias Biológicas ISSN: 0253-5688 [email protected] Centro Nacional de Investigaciones Científicas Cuba González, Lidice; Torres, Lino E.; Marrero, Karen; Reyes, Orlando; Rodríguez, Elaine; Rodríguez, Boris L. Clonaje y expresión heteróloga de un fragmento de la proteína CagA de Helicobacter pylori Revista CENIC. Ciencias Biológicas, vol. 41, 2010, pp. 1-11 Centro Nacional de Investigaciones Científicas Ciudad de La Habana, Cuba Disponible en: http://www.redalyc.org/articulo.oa?id=181220509022 Cómo citar el artículo Número completo Más información del artículo Página de la revista en redalyc.org Sistema de Información Científica Red de Revistas Científicas de América Latina, el Caribe, España y Portugal Proyecto académico sin fines de lucro, desarrollado bajo la iniciativa de acceso abierto
Transcript of Redalyc.Clonaje y expresión heteróloga de un fragmento de ... · El inserto escindido de 2,1 kb,...

Revista CENIC. Ciencias Biológicas
ISSN: 0253-5688
Centro Nacional de Investigaciones Científicas
Cuba
González, Lidice; Torres, Lino E.; Marrero, Karen; Reyes, Orlando; Rodríguez, Elaine; Rodríguez,
Boris L.
Clonaje y expresión heteróloga de un fragmento de la proteína CagA de Helicobacter pylori
Revista CENIC. Ciencias Biológicas, vol. 41, 2010, pp. 1-11
Centro Nacional de Investigaciones Científicas
Ciudad de La Habana, Cuba
Disponible en: http://www.redalyc.org/articulo.oa?id=181220509022
Cómo citar el artículo
Número completo
Más información del artículo
Página de la revista en redalyc.org
Sistema de Información Científica
Red de Revistas Científicas de América Latina, el Caribe, España y Portugal
Proyecto académico sin fines de lucro, desarrollado bajo la iniciativa de acceso abierto

1
Clonaje y expresión heteróloga de un fragmento de la proteína CagA de Helicobacter pylori Cloning and heterologous expression of a fragment of the Helicobacter pylori-CagA protein Lidice González, Lino E. Torres, Karen Marrero,* Orlando Reyes, Elaine Rodríguez, Boris L. Rodríguez. Departamentos de Microbiología e Inmunología y *Biología Molecular, Centro Nacional de Investigaciones Científicas, Apartado Postal 6412, Ciudad de La Habana, Cuba. Correo electrónico: [email protected]

2
Clonaje y expresión heteróloga de un fragmento de la proteína CagA de Helicobacter pylori Cloning and heterologous expression of a fragment of the Helicobacter pylori-CagA protein RESUMEN Introducción: Numerosos estudios confirman que la infección con cepas de Helicobacter pylori CagA-positivas aparece asociada al desarrollo de patologías gastroduodenales más severas, es por ello que la detección de anticuerpos anti-CagA adquiere una importancia significativa en la detección serológica de la infección con H. pylori. Este estudio fue llevado a cabo con el objetivo de expresar un fragmento recombinante de la proteína CagA con una cola de histidina (rCagA) que pueda ser empleado en un juego serológico rápido para detectar la infección con H. pylori y clasificar la misma en CagA-positiva o negativa. Materiales y Métodos: Un fragmento del gen cagA procedente de la cepa de referencia de H. pylori 17874 fue amplificado por PCR. El fragmento amplificado fue clonado en el vector de expresión pET22b(+) y esta construcción empleada para transformar la cepa de E.coli BL21 (DE3) y expresar el fragmento recombinante rCagA. La expresión fue inducida con IPTG y evaluada por SDS-PAGE. La técnica de Western blotting fue usada para determinar la inmunorreactividad de rCagA mediante el empleo de un antisuero de conejo específico Anti-CagA y un panel de sueros humanos CagA-positivos y negativos. Resultados y Discusión: Los resultados del experimento en SDS-PAGE demostraron que el sistema de expresión empleado con 0,1mM de IPTG y 4 h de inducción produjo eficientemente el rCagA con la talla molecular esperada de aproximadamente 80kDa. La banda del rCagA no se observo en ausencia de IPTG. La proteína expresada mostró una buena inmunorreactividad con el antisuero específico Anti-CagA, la cual fue fuerte con los sueros CagA-positivos y no se observó reactividad evidente con los sueros CagA-negativos empleados. Conclusiones: La proteína CagA recombinante obtenida parece ser útil para detectar y clasificar la infección con H. pylori por métodos serológicos. Palabras clave: Helicobacter pylori, proteína CagA recombinante, Juego serológico. ABSTRACT Introduction: Many evidences support that infection by CagA-positive Helicobacter pylori strains is related to the development of more severe gastroduodenal diseases, thus determination of anti-CagA antibodies acquire a relevant clinical significance in the serological detection of H. pylori infection. This study was carried out to express a recombinant fragment of CagA protein with an histidine tail (rCagA) that can be used in a rapid serological test to detect H. pylori infection and classify it as CagA positive or negative. Material and Methods: A fragment of the cagA gene was amplified by PCR from the H. pylori reference strain 17874. The prokaryote expression vector pET22b (+) plus the amplified cagA-DNA fragment was constructed, and used to transform the E.coli BL21 (DE3) strain to express the rCagA. The expression of the recombinant protein was induced by IPTG and examined by SDS-PAGE. Western blotting was used to determine the immunoreactivity of rCagA by using a specific rabbit antiserum and several CagA positive and negative human sera. Results and Discussion: SDS-PAGE experiments demonstrated that the expression system used efficiently produced the rCagA at the expected 80 kDa size with a 0.1 mM of IPTG and 4 hours of induction. No band of rCagA was observed in the absent of IPTG induction. The expressed CagA protein showed a good immunoreactivity with the Anti-CagA specific antiserum, that was higher with the

3
CagA positive sera, but none evident reaction was observed when the CagA negative human sera were used. Conclusion: The recombinant CagA protein obtained here since to be useful to detect and classify the H. pylori infection by serology. Keywords: Helicobacter pylori, CagA recombinant protein, Serological test. INTRODUCCIÓN Helicobacter pylori (H. pylori) es una bacteria Gram-negativa, espirilada y microaerofílica, que infecta alrededor del 50 % de la población mundial.1 Este microorganismo es el principal agente etiológico de varias enfermedades gastroduodenales como la gastritis, las úlceras pépticas, el cáncer gástrico, y el linfoma del tejido linfoide asociado a mucosa (Linfoma MALT).1 Debido a que se ha demostrado que este patógeno constituye un importante factor de riesgo en la aparición de cáncer gástrico y otras lesiones malignas fue clasificado en 1994 como la primera bacteria carcinógeno tipo I para el hombre por la Agencia de Investigaciones del Cáncer. 2
CagA es el primer antígeno de H. pylori que fue asociado a patologías gástricas. Esta proteína soluble de 120-145 kDa que no presenta ningún fragmento transmembranal, posee marcadas propiedades hidrofílicas con una región carboxilo terminal altamente variable. El marcado interés en esta proteína se debió inicialmente a su inmnunodominacia en pruebas serológicas, a pesar de que se expresa en bajas concentraciones por H. pylori.3 Esta toxina altera diversas vías de señalización en la célula epitelial gástrica con lo cual produce cambios en la morfología y fisiología de las mismas, lo cual se traduce en daño epitelial severo de la mucosa gástrica.4 Aproximadamente el 60 % de las cepas de H. pylori son cagA-positivas y numerosos estudios han correlacionado la infección de estas cepas con el riesgo de desarrollar úlcera péptica,5,6 así como cáncer gástrico.7 Mientras que por otra parte estudios en animales transgénicos han demostrado que definitivamente CagA es una oncoproteína, y que constituye además la primera oncoproteína bacteriana descrita.8 Por todas las razones anteriores, detectar las cepas CagA-positivas resulta de gran interés en pacientes que presentan trastornos gastroduodenales, de ahí que el objetivo de este trabajo fue obtener un fragmento recombinante de CagA que permita evaluar la seropositividad a la toxina en estos pacientes, lo que indica la presencia de cepas CagA positivas en ellos. MATERIALES Y MÉTODOS Obtención del fragmento de clonaje El fragmento a clonar se escogió dado que su secuencia aminoacídica estaba incluida en los fragmentos descritos en la literatura anterior y además, contenía motivos aminoacídicos que son inmunogénicos.9,10 El ácido desoxirribonucleico (ADN) de la cepa de referencia H. pylori 17874 fue empleado como molde para amplificar el fragmento del gen cagA, utilizando la técnica de reacción en cadena de la polimerasa (PCR). Los oligonucleótidos para la amplificación se sintetizaron en el Centro de Ingeniería Genética y Biotecnología (La Habana, Cuba) con las secuencias siguientes: oligonucleótido de cagA directo (5’- CATGCCATGGGGGATAACAGGCAAGCTTTTGA -3’), oligonucleótido de cagA reverso (5’- GAATTCTCGAGGTCGCTTTTTGC -3’). Los oligonucleótidos se diseñaron basados en la secuencia del gen de la cepa de referencia de H. pylori 26695, que está disponible en la base de datos del Genebank. Los mismos contenían además las secuencias de corte de las enzimas de restricción que se seleccionaron para realizar la construcción genética a clonar. El fragmento del gen cagA amplificado codifica para un polipéptido de una talla aproximada de 80 kDa.

4
Las reacciones de amplificación se llevaron a cabo de la manera siguiente: una primera etapa de desnaturalización a 94 °C durante un minuto, seguida de 40 ciclos que constaron de otra etapa de desnaturalización a la misma temperatura durante un minuto; un minuto de hibridación a 61 °C; una etapa de extensión a 72 °C durante dos minutos y una etapa final de extensión a 72 °C durante cinco minutos. Las reacciones se llevaron a cabo en el equipo de ciclos térmicos Mastercycler (Eppendorf, Alemania). La purificación del producto de PCR se realizó directamente de la reacción de amplificación mediante el empleo del High Pure PCR Product Purification Kit (Roche, Alemania). El resultado se analizó por electroforesis en gel de agarosa 1,6 % según lo descrito por Sambrook et al.11 Clonaje El plásmido pET22b(+) fue escogido por formar parte del sistema pET, el cual constituye uno de los sistemas más potentes de clonaje y expresión de proteínas recombinantes en E. coli. Este plásmido de 5,5 kb se purificó a partir de la cepa E. coli Mach1 (previamente transformada) mediante el empleo del Genopure Plasmid Midi Kit (Roche, Alemania) (Se empleó el procedimiento para un elevado número de copias). La purificación fue chequeada en electroforesis en gel de agarosa 0.8 % según lo descrito por Sambrook et al.11 El fragmento de ADN amplificado del gen cagA de 2,1 kb así como el vector pET22b(+) purificados fueron digeridos con las enzimas de restricción NcoI y XhoI (Roche, Alemania). Estas reacciones de digestión se llevaron a cabo en un volumen final de 30 µL y fueron empleadas 10U de cada una de las enzimas por cada 1 µg de ADN a digerir. La incubación de estas reacciones fue toda la noche a 37 ºC. Posteriormente se procedió a la purificación tanto del ADN plasmídico como del fragmento de PCR mediante el empleo del High Pure PCR Product Purification Kit (Roche, Alemania). Los productos de las digestiones se chequearon por electroforesis en gel de agarosa 0,8 %.11 El inserto escindido de 2,1 kb, codificante de un fragmento de la proteína CagA, se ligó al vector de expresión pET22b(+) digerido con las mismas enzimas, mediante el empleo de la enzima T4 ligasa (Roche, Alemania). Esta reacción de ligamiento se llevó a cabo en una proporción vector : banda de 1:4 y para ello se emplearon 4U de enzima por cada 1 µg de ADN a ligar. Esta construcción genética sitúa al fragmento de interés cuesta abajo del promotor T7, que tiene como inductor al IPTG. El fragmento recombinante clonado contiene además seis histidinas en el extremo carboxilo terminal, útiles para su purificación por cromatografía de afinidad de iones metálicos inmovilizados. Para realizar la transformación bacteriana, la cepa E. coli Mach1 (de elevada frecuencia de transformación) se cultivó en 5 mL de caldo LB,11 ajustado a pH 7,5 y se creció en zaranda orbital con termostato (Newbrunswik, USA) durante toda la noche a 37 °C y 200 rpm. La transformación bacteriana se realizó mediante electroporación según lo descrito por Dower et al.12 Posteriormente, se chequeó la presencia de los clones transformantes. A partir de estos clones, se purificó el ADN plasmídico mediante el empleo del High Pure Plasmid Isolation Kit (Roche, Alemania), lo cual fue chequeado en eletroforesis en gel de agarosa 0,8 % para ver si alguno de los clones contenía la construcción de interés. Una vez seleccionados los clones deseados se procedió a la purificación del vector con el inserto mediante el empleo del Genopure Plasmid Midi Kit (Roche, Alemania) para obtener una mayor concentración del plásmido de interés. Para corroborar que efectivamente estos clones seleccionados contenían el vector con el inserto se digirió el plásmido purificado por digestión NcoI-XhoI y con NcoI solamente y se chequearon las respectivas digestiones en gel de agarosa 0,8 %.

5
Expresión de la proteína recombinante Una vez obtenida una cantidad de vector con inserto suficiente se procedió a la transformación de la cepa de expresión E. coli BL21(DE3) de la misma manera que fue descrita para la cepa Mach1. Esta cepa fue seleccionada por ser una de las cepas más empleadas para expresar proteínas recombinantes clonadas en el sistema pET. Esta cepa transformada con la construcción de interés, se usó para expresar el polipéptido de CagA, la cual fue crecida en caldo LBA a 37 °C y 200 rpm hasta una DO600nm de aproximadamente 0.5 a partir de la cual se indujo la expresión por adición de IPTG (isopropil-β-D-tiogalactopiranósido) a una concentración final de 0,1 mM e incubación de 4 h adicionales. Para la colección de la biomasa bacteriana se tomó una alícuota de 1 mL y se centrifugó cinco min a 12 000 rpm. Por otra parte se colectó también una alícuota de 1 mL de otro cultivo de esta cepa transformada (que fue incubado bajo similares condiciones que el anterior) en el cual no fue inducida la expresión. La evaluación de la expresión del fragmento de CagA se realizó por análisis del perfil proteico total de la cepa E. coli BL21(DE3) transformada, mediante electroforesis desnaturalizante en geles de poliacrilamida (SDS-PAGE) según Laemmli.13 Los pellets celulares se resuspendieron en disolución estabilizadora de fosfato salino (PBS), de forma que la concentración celular fuera equivalente para todas las muestras, se mezcló con igual volumen de disolución reguladora de SDS y se hirvió durante diez minutos. Las muestras se separaron en geles de acrilamida al 12 %, en presencia de disolución reguladora tris-glicina y los mismos fueron teñidos con azul de Coomasie. Para comprobar por inmunodetección la presencia del fragmento de CagA, se empleó la técnica del Western Blot (WB), que se desarrolló según el procedimiento descrito por Towbin y col.14 Se utilizaron membranas de nitrocelulosa HybondTM-C (Amersham, Reino Unido). Las membranas se incubaron con un suero que presenta inmunorreacción positiva a la proteína CagA (el suero se diluyó 1 : 100 en PBS con tween-20 al 0,05 %) durante toda la noche con agitación y a temperatura ambiente. Luego, las membranas se lavaron y se incubaron durante 3 h con un anticuerpo monoclonal específico para la cadena pesada de la IgG humana conjugado a peroxidasa (CIGB, La Habana, Cuba), diluido 1 : 1 000 en PBS con tween-20 0,05 % y leche descremada al 1 %. Finalmente, el revelado se efectuó con una disolución de diaminobenzidina (0,24 %) disuelta en disolución estabilizadora de Tris (20 mmol · L–1, pH 7,6), a la que se añadieron 40 μL de peróxido de hidrógeno 30 %. Para corroborar que el fragmento obtenido era realmente un fragmento de la proteína CagA se realizó un WB en el que una de las membranas de nitrocelulosa que contenía el extracto celular de un cultivo inducido se incubó con un antisuero específico anti-CagA de conejo. Este antisuero específico había sido generado previamente a partir de un fragmento recombinante de esta proteína obtenido por otro grupo de trabajo.15 La técnica del WB fue realizada tal y como fue descrita con anterioridad con la variante en este caso de que como el antisuero es de conejo, el anticuerpo de reconocimiento empleado fue un anticuerpo policlonal de conejo específico para la IgG molécula completa conjugado a peroxidasa (SIGMA, USA). Para evaluar preliminarmente las propiedades de inmunorreacción del péptido recombinante de CagA, se prepararon membranas con el extracto celular del cultivo inducido, así como con el extracto celular de un cultivo sin inducir que se creció en similares condiciones que el anterior, y se analizaron por WB 15 sueros H. pylori-positivos/CagA-positivos (Hp+/CagA+) y 15 sueros Hp+/CagA-. Estos sueros ya habían

6
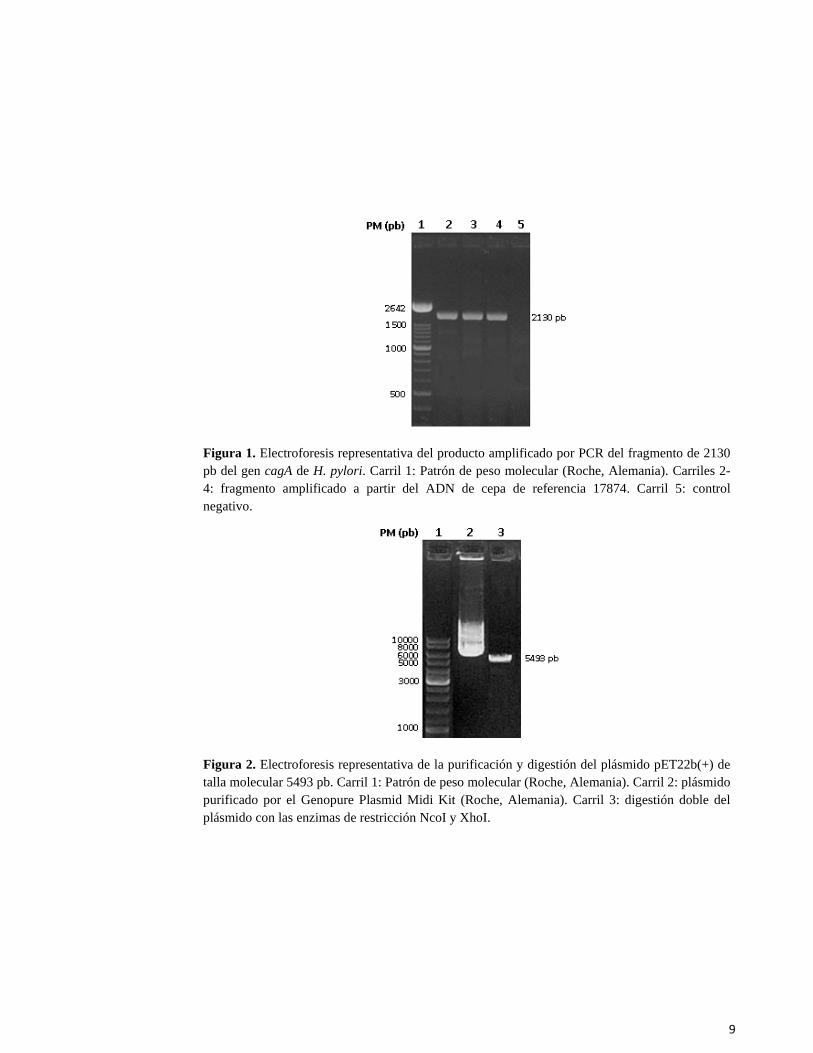
sido clasificados previamente mediante un WB que empleó el extracto proteico de una cepa de H. pylori CagA-positiva.16 La técnica del WB fue realizada tal y como fue descrita con anterioridad. RESULTADOS Y DISCUSIÓN Obtención y clonaje de la construcción genética El fragmento seleccionado del gen cagA para el clonaje, que fue amplificado por PCR y chequeado por electroforesis en geles de agarosa 1,6 %, mostró la talla molecular esperada de 2130 pb, evidenciándose la efectividad de los oligonucleótidos diseñados (Fig. 1). Mientras, el plásmido pET22b(+) purificado y linealizado por digestión con las enzimas de restricción NcoI y XhoI también mostró la talla esperada de 5493 pb (Fig. 2). Una vez obtenidos y chequeados los materiales genéticos de partida, se procedió al ligamiento de los mismos para crear la construcción genética de interés que iba a ser introducida en la cepa de multiplicación Mach1. La transformación inicial del producto de ligamiento en la cepa Mach1 permitió la obtención de 28 clones transformantes de los cuales una vez purificado su ADN plasmídico se seleccionó solo un posible clon que parecía haber incorporado la construcción genética de interés (Fig. 3). La digestión doble del plásmido purificado a partir de este único clon transformante con las enzimas de restricción NcoI y XhoI produjo fragmentos de talla 5,5 y 2,1 kb. Estos fragmentos corresponden con las tallas tanto del fragmento de interés amplificado por PCR como del vector pET22b(+) linealizado. Este resultado nos indicó que al parecer este único clon seleccionado contenía la construcción genética de interés. Por otra parte, la digestión simple de este mismo plásmido purificado, produjo un fragmento de talla molecular 7,6 kb, que corresponde con la talla esperada del vector más el fragmento amplificado por PCR (Fig 4), lo cual refuerza lo anteriormente expuesto. Expresión y evaluación de la proteína recombinante Una vez seleccionado el clon que contenía la construcción genética de interés fue transformada la cepa de expresión BL21(DE3), obteniéndose un buen rendimiento de transformantes con más de 50 clones por cada placa de cultivo. De esta forma se procedió a la inducción de la expresión con IPTG. La evaluación de la expresión se realizó por SDS-PAGE donde se detectó la aparición de una banda de gran intensidad y grosor de aproximadamente 80 kDa, la cual se correspondía con la talla teórica esperada de la proteína recombinante según el fragmento de DNA amplificado (Fig. 5, Panel A). Por su parte, en el caso del cultivo sin inducir no se observó dicha banda proteica (Fig. 5, Panel A). Este resultado es de gran importancia, ya que nos indicó que el sistema de expresión utilizado producía una proteína recombinante como la talla esperada y además en grandes cantidades. El próximo paso en el estudio fue comprobar que esta proteína recombinante era reconocida en principio por el suero de un paciente Hp+/CagA+ que contiene anticuerpos anti-CagA, mediante la técnica de WB. El resultado de este experimento mostró la presencia de una banda de inmunorreacción de gran intensidad que también coincidió con la talla molecular teórica del fragmento recombinante expresado (Fig. 5, Panel B). Este hecho es el segundo factor de gran importancia en el estudio, ya que nos sugirió que además de obtenerse en grandes cantidades la proteína recombinante presentaba muy buenas propiedades para el diagnóstico serológico, dada la fuerte inmunorreacción ante un suero CagA-positivo, así como una débil inmunorreacción ante las proteínas de la cepa de E. coli seleccionada para producir dicho fragmento recombinante.

7
Por otra parte, para comprobar que se trataba de un fragmento de la proteína CagA, se realizó otro experimento en el cual se incubó una membrana que contenía el extracto de un cultivo inducido con un antisuero específico anti-CagA, generado a partir de un fragmento recombinante de esta proteína obtenido por otro grupo de trabajo.15 Este experimento, donde se obtuvo también una fuerte banda de inmunorreacción en la talla esperada, permitió confirmar que la proteína recombinante obtenida se trataba en efecto de un fragmento de la proteína CagA (Fig. 5, Panel C). La evaluación preliminar de la especificidad del péptido recombinante se realizó con 15 sueros de pacientes Hp+/CagA+ y 15 sueros de pacientes Hp+/CagA- mediante el empleo de membranas preparadas tanto con el extracto celular de cultivos inducidos y sin inducir. En las membranas preparadas con el cultivo inducido, todos los sueros clasificados como seropositivos a CagA presentaron una fuerte inmunorreacción con el fragmento recombinante (Fig. 6, Panel A), la cual no fue observada ni con los sueros negativos a CagA incubados con esta membrana (Fig. 6, Panel A), ni con los sueros CagA+ o CagA- incubados con la membrana que contenía el extracto del cultivo sin inducir (Fig. 6, Panel B). Sin embargo, en algunos de los sueros de los pacientes se observó una banda de inmunorreacción débil en la zona que se corresponde con el peso molecular del CagA recombinante. Esta reacción pudiera deberse a uno de los factores siguientes: la presencia de una proteína de E. coli de talla molecular muy similar al fragmento recombinante que inmunorreacciona débilmente con algunos de los sueros, o la positividad débil de alguno de los sueros a CagA que no fue detectada cuando estos sueros fueron clasificados previamente. Este resultado será elucidado una vez que el rCagA sea purificado y se proceda nuevamente a la evaluación de un panel más amplio de sueros. Todos estos resultados sugieren que el fragmento clonado de CagA presenta muy buenas propiedades para clasificar los sueros de pacientes dispépticos como seropositivos o no a esta toxina, lo que debe ratificarse una vez que se purifique la proteína y se evalúe con un número mayor de sueros. En Cuba, sería de gran importancia contar con una herramienta para este inmunodiagnóstico, pues en un estudio previo se detectó una elevada seropositividad a la toxina CagA en pacientes dispépticos cubanos.17
CONCLUSIONES La construcción pET22b(+)-CagA es muy eficiente para la expresión heteróloga en E. coli de un fragmento recombinante de la proteína CagA de H. pylori. El fragmento recombinante de CagA parece tener buenas propiedades para el inmunodiagnóstico, lo cual debe validarse con la proteína purificada y la evaluación de un panel de sueros más amplio. REFERENCIAS BIBLIOGRÁFICAS
1. Go MF. Review article: natural history and epidemiology of Helicobacter pylori infection. Aliment Pharmacol Ther. 2002;16:3-15.
2. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Carcinog Risks Hum. 1994;61:1-241.
3. Tummuru MK, Cover TL, Blaser MJ. Mutation of the cytotoxin-associated cagA gene does not affect the vacuolating cytotoxin activity of Helicobacter pylori. Infect Immun.1994;2:2609-2613.

8
4. Hatakeyama M. Linking epithelial polarity and carcinogenesis by multitasking Helicobacter pylori virulence factor CagA. Oncogene. 2008;27:7047-7054.
5. Lamarque D, Gilbert T, Roudot-Thoraval F, Deforges L, Chaumette MT, Delchier JC. Seroprevalence of eight Helicobacter pylori antigens among 182 patients with peptic ulcer, MALT gastric lymphoma or non-ulcer dyspepsia. Higher rate of seroreactivity against CagA and 35-kDa antigens in patients with peptic ulcer originating from Europe and Africa. Eur J Gastroenterol Hepatol. 1999;11:721-726.
6. Nomura AM, Perez-Perez GI, Lee J, Stemmermann G, Blaser MJ. Relation between Helicobacter pylori cagA status and risk of peptic ulcer disease. Am J Epidemiol. 2002;155:1054-1059.
7. Wu AH, Crabtree JE, Bernstein L, Hawtin P, Cockburn M, Tseng CC, et al. Role of Helicobacter pylori CagA+ strains and risk of adenocarcinoma of the stomach and esophagus. Int J Cancer. 2003;103:815-821.
8. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105 (3):1003-8.
9. Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci. 1993;90:5791-5795.
10. Tummuru MK, Cover TL, Blaser MJ Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun. 1993;61:1799-1809.
11. Sambrok J, Fritsch EF, Maniatis T. Molecular Cloning: a laboratory manual, Cold Spring Harbor, N.Y., USA: Cold Spring Harbor Laboratory Press: 1989.
12. Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988;16:6127.
13. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680-685.
14. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of protein from polyacrilamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci. 1979;76:4350-4354.
15. Tummuru MK, Cover TL, Blaser MT. Cloning and Expression of a High-Molecular-Mass Major Antigen of Helicobacter pylori: Evidence of Linkage to Cytotoxin Production. Infec Immun. 1993;61(5):1799-809.
16. Torres LE, Bermúdez L, Roblejo Y, Moreno A, Samada M, Cansino J et al. Desarrollo de un método serológico propio para el diagnóstico de la infección por Helicobacter pylori y su comparación con dos juegos comerciales. Revista CENIC Ciencias Biológicas. 2008;39:115-120.
17. Valmaseda T, Gisbert JP, Paniagua M, Pajares JM. Helicobacter pylori CagA antibodies in various gastroduodenal diseases from 2 different populations. Med Clin (Barc). 2002;118:90-93.
9
Figura 1. Electroforesis representativa del producto amplificado por PCR del fragmento de 2130 pb del gen cagA de H. pylori. Carril 1: Patrón de peso molecular (Roche, Alemania). Carriles 2- 4: fragmento amplificado a partir del ADN de cepa de referencia 17874. Carril 5: control negativo.
Figura 2. Electroforesis representativa de la purificación y digestión del plásmido pET22b(+) de talla molecular 5493 pb. Carril 1: Patrón de peso molecular (Roche, Alemania). Carril 2: plásmido purificado por el Genopure Plasmid Midi Kit (Roche, Alemania). Carril 3: digestión doble del plásmido con las enzimas de restricción NcoI y XhoI.

10
Figura 4. Electroforesis representativa de la digestión de la construcción genética pET22b(+)-CagA purificada de la cepa Mach1. Carril 1: plásmido pET22b(+) digerido con las enzima NcoI. Carril 2: digestión doble de la construcción genética pET22b(+)-CagA con las enzimas NcoI y XhoI. Carril 3: fragmento amplificado por PCR del gen cagA de H. pylori. Carril 4: Patrón de peso molecular (Roche, Alemania). Carril 5: digestión simple de la construcción genética pET22b(+)-CagA con la enzima NcoI.
Figura 3. Electroforesis representativa de la purificación del ADN plasmídico de los clones transformantes de la cepa E. coli Mach1. Carriles 1-10: ADN plasmídico purificado a partir de 10 clones transformantes mediante el High Pure Plasmid Isolation Kit (Roche, Alemania). Carril 11: Patrón de peso molecular (Roche, Alemania). Carril 12: plásmido digerido con las enzimas de restricción NcoI y XhoI y purificado con el Genopure Plasmid Midi Kit (Roche, Alemania).

11
Figura 5. Gel de SDS-PAGE 12 % y WB de la cepa E. coli BL21 (DE3) transformada con él plásmido pET22b(+)-CagA sin inducir e inducida con IPTG. Panel A: SDS-PAGE de los extractos proteicos de la cepa de E. coli BL21 (DE3). Carril 1: Patrón de peso molecular (Promega, USA). Carril 2: Clon transformado inducido con IPTG. Carril 3: Clon transformado sin inducir. Panel B: SDS-PAGE y WB. Carril 1: Patrón de peso molecular (Promega, USA). Carril 2: Clon transformado inducido con IPTG e incubado con un suero de paciente dispéptico CagA-positivo. Carril 3: Clon transformado sin inducir e incubado con el mismo suero CagA-positivo. Panel C: SDS-PAGE y WB. Carril 1: Patrón de peso molecular (Promega, USA). Carril 2: Clon transformado sin inducir e incubado con antisuero de conejo específico anti-CagA. Carril 3: Clon transformado inducido con IPTG e incubado con el mismo antisuero específico anti-CagA.
Figura 6. WB del extracto proteico de E. coli BL21 (DE3) transformada con pET22b(+)-CagA inducida y sin inducir con IPTG e incubadas con sueros de pacientes dispépticos Hp+/CagA+ y HP+/CagA-. Panel A: Extracto celular del clon BL21 (DE3) transformado con pET22b(+)-CagA inducido con IPTG. Carril 1: Incubado con suero control Hp+/CagA+. Carriles 2-8: Incubados con sueros Hp+/CagA+. Carriles 9-16: Incubados con sueros Hp+/CagA-. Carril 17: Incubado con suero Hp- (control negativo). Panel B: Clon BL21 (DE3) transformado con pET22b(+)-CagA sin inducir. Carril 1: Incubado con suero control Hp+/CagA+. Carriles 2-8: Incubados con sueros Hp+/CagA+. Carriles 9-16: Incubados con sueros Hp+/CagA-. Carril 17: Incubado con suero Hp- (control negativo).


















