Anemia Ferropenica
43
Anemia Ferropénica
-
Upload
magaly-marquez -
Category
Documents
-
view
17 -
download
0
description
La frecuencia de la anemia ferropénica depende de varios aspectos básicos del metabolismo del hierro y de la nutrición.El niño debe absorber alrededor de 0.8 a 1 mg de Hierro al díaEl organismo del recién nacido contiene aproximadamente 0,5 g de hierro, mientras que el del adulto tiene alrededor de 5 g.La absorción de 0.8 mg debe ser durante los primeros 15 años.
Transcript of Anemia Ferropenica

Anemia Ferropénica

Anemia Ferropénica
La frecuencia de la anemia ferropénica depende de varios aspectos básicos del metabolismo del hierro y de la nutrición.
El niño debe absorber alrededor de 0.8 a 1 mg de Hierro al día
El organismo del recién nacido contiene aproximadamente 0,5 g de hierro, mientras que el del adulto tiene alrededor de 5 g.
La absorción de 0.8 mg debe ser durante los primeros 15 años.

Se cree que se absorbe alrededor del 10% del hierro de la dieta, por lo que para una nutrición óptima la dieta diaria debe contener entre 8 y 10 mg de este elemento.
El hierro se absorbe en la zona proximal del intestino delgado
Los niños alimentados con lactancia materna necesitan menos hierro de los demás alimentos.
El lactante se encuentra en una situación precaria en relación con el hierro. Si la dieta es inadecuada o sufren pérdidas de sangre externas, la anemia se desarrollará con rapidez.
Concepto
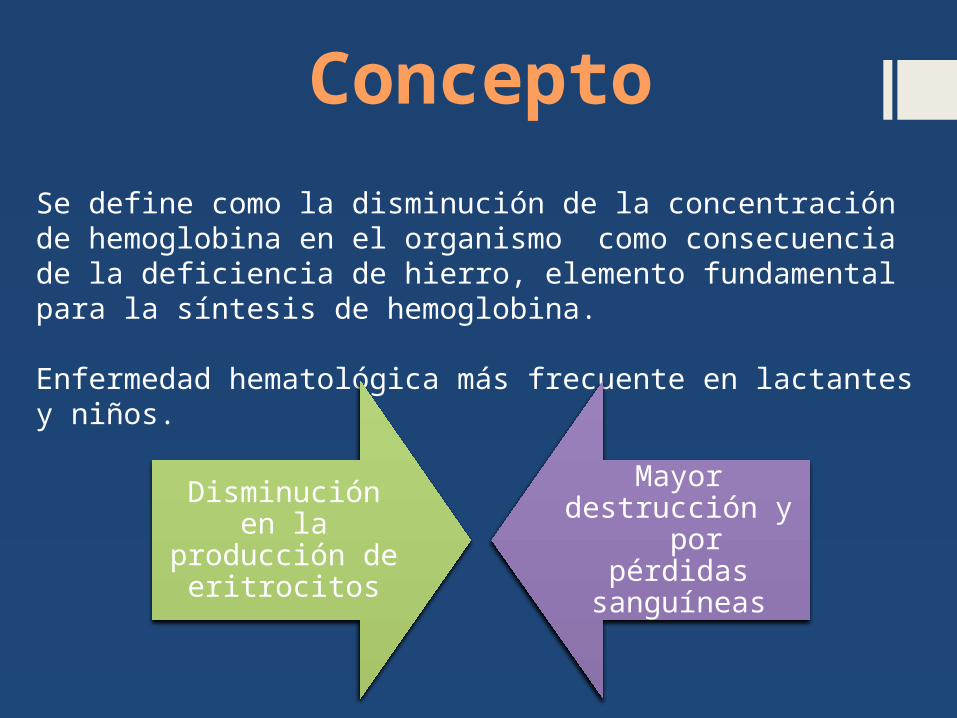
Se define como la disminución de la concentración de hemoglobina en el organismo como consecuencia de la deficiencia de hierro, elemento fundamental para la síntesis de hemoglobina.
Enfermedad hematológica más frecuente en lactantes y niños.
Disminución en la producci
ón de eritrocito
s
Mayor destrucción y por pérdidas sanguíne
as

ETIOLOGÍA
Disminución nutricional del aporte de hierro (es la más común)
Alteración de la digestión o de la absorción del hierro
Pérdidas sanguíneas digestivas. Enterocolitis provocada por leche de vaca, parasitosis intestinal (giardia lamblia), divertículos de Meckel

Fisiopatología
RN tiene reservas adecuadas de hierro hasta los 4-6 meses de edad
Esta reserva proviene del aporte de hierro en la VIU
El hierro de la madre es incorporado por el feto durante el 3er trimestre

En la mayoría de los casos la anemia ferropénica en el lactante y en la primera infancia está determinada por una dieta insuficiente o mal balanceada.
El defecto habitual es la introducción tardía o el rechazo de alimentos ricos en hierro en la dieta del lactante
La incorporación temprana de la leche de vaca (antes de los seis meses de vida) es otro factor causal de importancia

Las pérdidas de hierro por descamación se suplen perfectamente a través de la dieta.
La prevalencia de anemia ferropénica es elevada especialmente si los mecanismos de absorción no funcionan correctamente.

La dosis total a administrar (para corregir la anemia y reponer los depó- sitos) se calculará de acuerdo a la siguiente fórmula
Cuadro Clínico

DIAGNÓSTICO
Historia clínica
Laboratorio:
Hb
Hto
VCM
CHCM
Recuento de reticulocitos
Recuento de plaquetas
Recuento leucocitario

TratamientoUso de sulfato ferroso
4.6 mg/kg/día (2-3 mg) durante 4 meses en promedio
Fumarato ferroso
3-6 mg/kg/día
Hierro Dextrán
Niños menores de 5 kg: 25 mgNiños entre 5 y 10 años: 50 mgNiños mayores de 10 kg: Hasta 100 mg

La dosis total a administrar (para corregir la anemia y reponer los depó- sitos) se calculará de acuerdo a la siguiente fórmula
Vía Parenteral

PREVENCIÓN PRIMARIA
Lactancia materna exclusiva (como mínimo 6 meses)
Uso de leche artificial (que contengan hierro)
A los niños nacidos pretérmino, de bajo peso, se les debe dar un suplemento de hierro oral en el primer año.
Niños menores de 6 meses:

Niños mayores de 6 meses:
Alimentados exclusivamente con lactancia materna hasta los 6 meses: asegurar la ingesta de 1mg/Kg/día de Fe en los alimentos de continuación o suplementar esta cantidad con Fe oral en gotas
-Al incorporar alimentos de continuación sólidos, garantizar el consumo de Fe teniendo en cuenta las siguientes indicaciones:
• Dos tomas diarias de cereales enriquecidos con hierro cubren los requerimientos necesarios (B).
• Los alimentos ricos en vitamina C (frutas, verduras, zumos...) mejoran la absorción de hierro.

PREVENCIÓN SECUNDARIA
Su objetivo es el diagnóstico precoz mediante un cribado analítico (hemoglobina, hematocrito y ferritina) y el tratamiento de la deficiencia de hierro.
Niños menores de 5 años:
Niños pertenecientes a familias en situación de desigualdad socioeconómica y cultural:
- realizar controles entre los 9 y 12 meses de edad, 6 meses después y anualmente hasta los 5 años.

Se aconseja el cribado de anemia en la población infantil en situaciones de riesgo:
• Niños nacidos pretérmino y de bajo peso, con sospecha carencial.
• Niños alimentados exclusivamente con lactancia materna, después de los 6 meses de edad.
• Niños en situaciones especiales (procesos infecciosos, desórdenes inflamatorios, tratamientos que interfieren la absorción, dietas restrictivas, pérdidas importantes de sangre por accidentes, o cirugía...) (B).
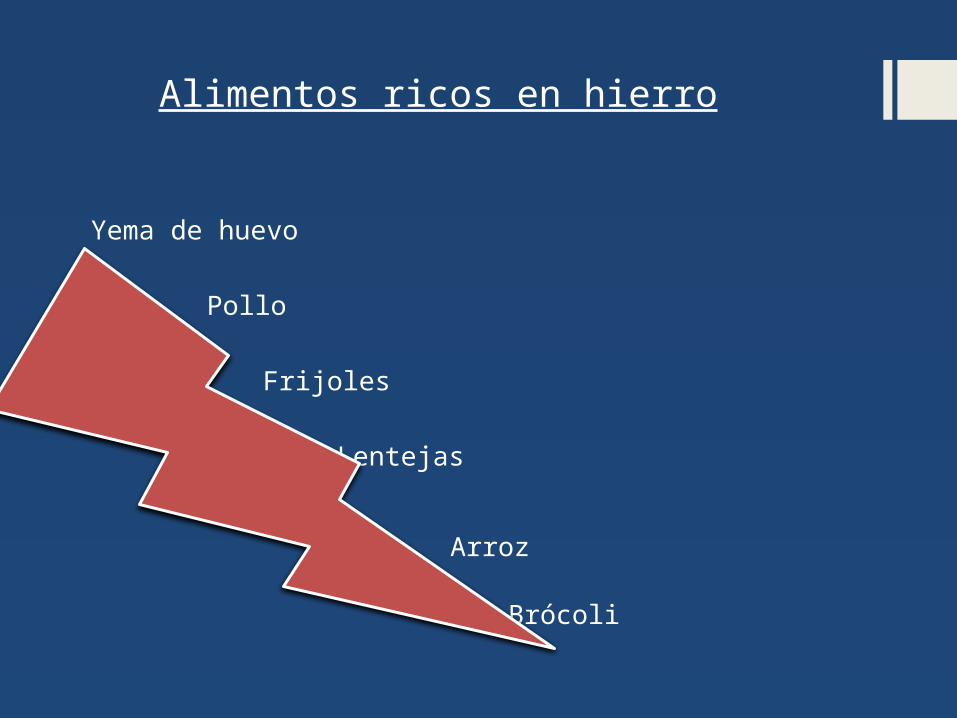
Alimentos ricos en hierro
Yema de huevo
Pollo
Frijoles
Lentejas
Arroz
Brócoli

Anemia hemolitica

Características generales
El termino anemia hemolítica agrupa a un conjunto de trastornos en los que se produce una destrucción acelerada de los hematíes
(HEMOLISIS), con disminución de su supervivencia (< 120 días)
MECANISMO COMPESATORIO
Aumento de la eritropoyesis (para garantizar el adecuado transporte de oxigeno)
> Destrucción y < producción
ANEMIA

Clasificación de la Anemia Hemolítica
MECANISMO
CORPUSCULARES O INTRINSECAS (AH CONGENITA)
EXTRACORPUSCULARES O EXTRINSECAS
(AH ADQUIRIDA)

Clasificación de la Anemia Hemolítica
SITIO DE LA HEMOLISIS
DURACION
o EXTRAVASCULAR (BAZO)
o INTRAVASCULAR
o AGUDAS (suelen ser intravasculares y cursan con hemoglobinuria, anemia e ictericia)
o CRONICAS( suelen se extravasculares y cursan con ictericia esplenomegalia y colelolitiasis)

MECANISMOS DE DESTRUCCIÓN DE LOS HEMATÍES
Normalmente la HEMOCATERESIS de los eritrocitos está relacionada con la edad celular (Deterioro de sistemas enzimáticos, disminución del ATP) y se da en el Sistema Fagocítico Mononuclear (SFM) = BAZO
Anemia Hemolítica
Extravascular Intravascular

Anemia hemolítica congénita

Trastorno de la membrana eritrocitaria
La membrana del hematíe está formada por una doble capa fosfolipídica y las proteínas integrales y estructurales que constituyen el citoesqueleto
defectos en la composición proteínica = pérdida de la forma del hematíe = reducción de vida del eritrocito

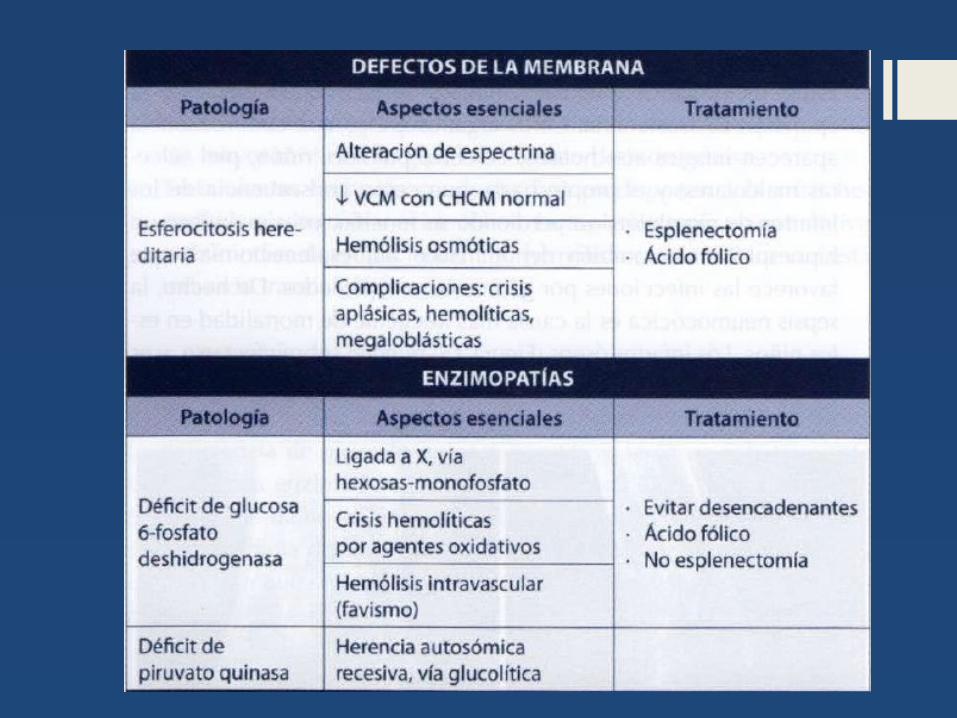
Esferocitosis hereditaria o enfermedad de Minkowski-Chauffard
AHC POR DEFECTO DE LA MEMBRANA
Forma leve Pueden permanecer asintomáticos, sin anemia, con hemólisis mínima, detectándose con motivo de estudios familiares o tras presentar una litiasis biliar en la edad adulta
Forma moderada La forma más frecuente de presentación se detecta en los primeros años de vida con anemia, esplenomegalia e ictericia que, ocasionalmente, requiere alguna transfusión.
Forma grave Muy poco frecuentes son los pacientes con anemia y hemólisis graves, con requerimientos transfusionales frecuentes
DEFINICIÓN: - Causa mas común de AHC- Esferocitos. - autosómico dominante (80%)- Frecuencia de 1 en 5,000DIAGNÓSTICO:
o La morfología presencia de esferocitos.o En el hemograma: VCM disminuido, CHCM
aumentada.o Prueba de la fragilidad osmótica: consiste en colocar
los hematíes del paciente en un medio hipoosmolar, y observar cómo se produce la hemolisis por la alteración de la permeabilidad citada de la membrana del hematíe.
o LDL y BI aumentadas.o Electroforesis en gel de poliacrilamida (PAGE):
Permite determinar la presencia o ausencia de proteínas de membrana de GR

AHC POR DEFECTO DE LA MEMBRANA
Eliptocitosis congénita
FISIOPATOLOGÍAConsiste en un defecto de la espectrina, que ocasiona una forma elíptica anormal del hematíe, pero no se acompaña de fragilidad osmótica lo que conducir a cambios esqueléticos que pueden hacer que la célula cambie a la forma elíptica
DEFINICIÓN: - eritrocitos ovalados o elípticos, - herencia autosómica dominante - 1 de 4000 o 5000 habitantes.
Clínica y diagnostico:o Asintomática (87% de los casos) y el resto puede cursar con
anemia presente.o En el hemograma se encuentra la presencia de eliptocitos
(eritrocitos con relación diámetro longitudinal/ transversal > 1) en porcentajes mayores al 12%.
o 10 a 15% de pacientes la hemólisis es considerablemente mayor.o El diagnóstico definitivo se hace con el análisis del ADN para
identificar el tipo de mutación
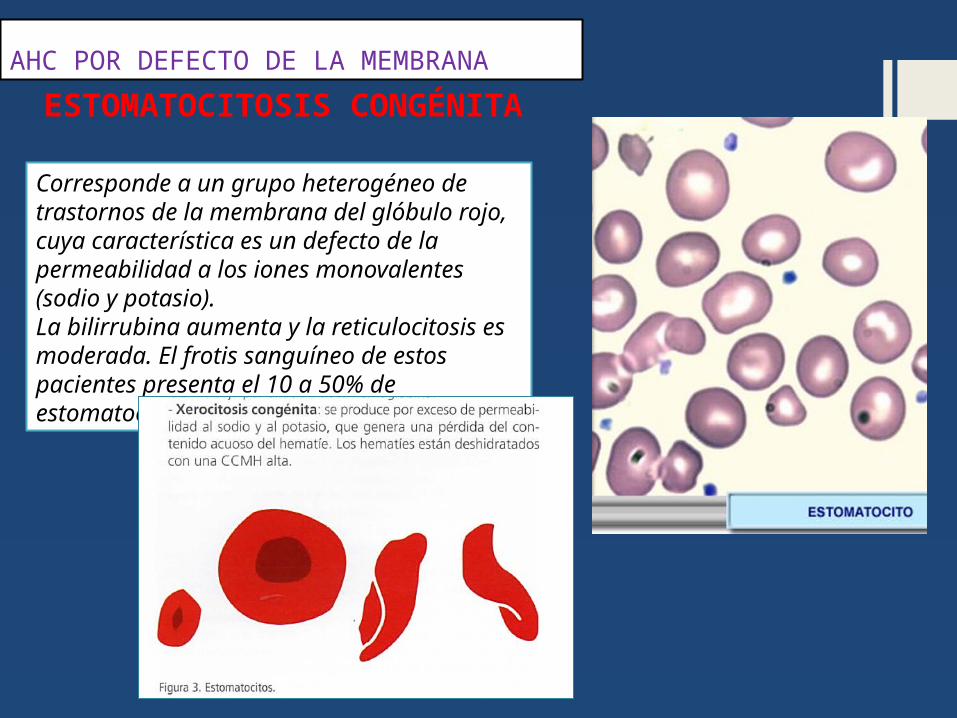
AHC POR DEFECTO DE LA MEMBRANA
Corresponde a un grupo heterogéneo de trastornos de la membrana del glóbulo rojo, cuya característica es un defecto de la permeabilidad a los iones monovalentes (sodio y potasio).La bilirrubina aumenta y la reticulocitosis es moderada. El frotis sanguíneo de estos pacientes presenta el 10 a 50% de estomatocitos.
ESTOMATOCITOSIS CONGÉNITA

AH POR DEFICIENCIAS ENZIMÁTICAS
Trastornos de la vía hexosa-monofosfato
Deficiencia de glucosa-6-fosfato deshidrogenasa.
- Es la causa más frecuente de anemia hemolítica
- herencia ligada al cromosoma X - Prevalencia relacionada con áreas de
paludismo endémico. - Esta enzima cataliza la primera reacción de
la vía de las pentosas fosfato y su función principal es proteger el eritrocito de agentes oxidantes.
PATOGENÍA: La utilidad G6PD es la generación de NADPH, cuya finalidad es reducir el glutatión, que a su vez evita la oxidación de los grupos sulfhidrilos de la Hb. La oxidación de los grupos sulfhidrilos produce metahemoglobina, que precipita en el interior del hematíe ocasionando los denominados cuerpos de Heinz, que ocasiona hemolisis intravascular y extravascular por lesión de la membrana del hematíe.

MANIFESTACIONES CLÍNICAS: desde casos asintomáticos o con hemolisis compensada hasta procesos hemolíticos neonatales graves. En situaciones especiales, se puede producir un incremento de la hemolisis (crisis hemolítica), que generalmente son secundarias a infecciones.
AHC POR DEFICIENCIAS ENZIMÁTICASTrastornos de la vía hexosa-monofosfato
Deficiencia de glucosa-6-fosfato deshidrogenasa.
DIAGNÓSTICO: o Hematólogicos: aumento del HCM,
cuerpos de Heinz (hemoglobina desnaturalizada que precipita en el interior del eritrocito.
o Bioquímicas. aumento de bilirrubina plasmática y uroblinógeno urinario y fecal.
o Medir la actividad enzimática y establecer el tipo de mutación.

Trastornos de la vía glucolítica o de Embden-Meyerhof
AHC POR DEFICIENCIAS ENZIMÁTICAS
Deficiencia De Piruvato-quinasa
- causa más frecuente de trastornos enzimáticos de la vía glucolítica (90 %)
PATOGENIALas enzimopatías del metabolismo glucolítico alteran la capacidad energética del eritrocito, dificultando la formación o utilización del ATP.Cuando disminuye la capacidad energética del eritrocito éste envejece prematuramente y es eliminado de la circulación sanguínea por el sistema fagocítico mononuclear (SFM).
MANIFESTACIONES CLÍNICASEl cuadro hemolítico se puede presentar desde el periodo neonatal o en la primera década de la vida y sus características son parecidas a las del cuadro clínico de la esferocitosis hereditaria, excepto por la presencia de esferocitos circulantes y fragilidad osmótica normal
DIAGNÓSTICOHemoglobina entre 6 a 12 g/dl, reticulocitosis, moderada macrocitosis (VCM: 98-105 fl), disminución de la vida media eritrocitaria, equinocitosis ausencia de esferocitos circulantes y fragilidad osmótica normal.Determinacion de actividad enzimatiza
En la mayoría de los casos de AHC por defecto de la membrana el tratamiento a seguir es la esplecnotomía, pues el objetivo es eliminar el lugar donde se destruyen los eritrocitos.
Como en todas las anemias hemolíticas, administrar ácido fólico para prevenir las crisis megaloblásticas.
Tratamiento

AHC POR HEMOGLOBINOPATIAS
Son trastornos de la hemoglobina
Síntesis de una cadena de globina estructuralmente anormal
Hemoglobinopatías estructurales
Ausencia o bien disminución en la síntesis de una cadena normal
Síndromes talasémicos

AHC POR HEMOGLOBINOPATIAS
A partir de los 4 años (Hb <de 8 gr/ L). Las crisis
vaso oclusivas afectan al pulmón, riñón y tejido
óseo; caracterizándose por dolor intenso en los
territorios afectados.
Hemoglobinopatías estructuralesAnemia Drepanocitica (Hemoglobinopatía S)
• Se debe a la sustitución del ácido glutámico en posición 6 de la cadena beta por
valina, con lo cual hay sustitución de adenina por timina en el código ADN. Las
moléculas de desoxihemoglobina S se agregan ordenadamente, formando
microtúbulos en forma helicoidal; debido a ésto surge la forma de hoz del
eritrocito. En el desarrollo clínico se pueden considerara tres fases
Fase Estacionaria
1-4 años. Tiene manifestaciones
clínicas de un cuadro hemolítico
moderado o intenso: anemia, ictericia y retraso del crecimiento óseo y gonadal.
Fase de Expresividad Aguda Fase de Expresividad Crónica
Se presenta en la adolescencia y en la edad adulta, afectando el
crecimiento y el desarrollo corporal así como el sistema
nervioso central, cardiovascular, pulmonar, gastro intestinal y
renal. Otra complicación relativamente frecuente en esta fase es la presencia de úlceras
maleolares de evolución tórpida.

AHC POR HEMOGLOBINOPATIAS
DIAGNÓSTICO Hemograma: muestra una anemia
normocítica o ligeramente macrocítica con hemoglobina entre 7 y 9 gr/dl, acompañada de reticulocitosis. El frotis evidencia la presencia de drepanocitos y dianocitos.
Electroforesis de Hemoglobinas a pH alcalino: Es el procedimiento diagnóstico más usado.
Hemoglobinopatías estructuralesTratamiento curativo
o Folato: 1 - 5 mg/ día en forma permanente.
o Transfusiones sanguíneas: solo cuando sea necesario, pues al aumentar la viscosidad sanguínea se incrementa el riesgo de hemólisis.
o Hidratación adecuada: cuyo objetivo es evitar la hiperviscosidad sanguínea.
o Analgésicos: de acuerdo a la intensidad del dolor.
o Agentes antidrepanociticos: Hidroxiurea, cuyo objetivo es incrementar la concentración de hemoglobina fetal (HbF).
o Transplante de Médula Ósea: Este procedimiento se debe reservar para los casos de mal pronóstico o incompatibles con una calidad de vida mínimamente aceptables.
o Tratamiento preventivo: Evitar todas aquellas situaciones que desencadenen las crisis vaso oclusivas: infecciones, acidosis, hipoxemia y exposición al frío .

AHC POR HEMOGLOBINOPATIAS
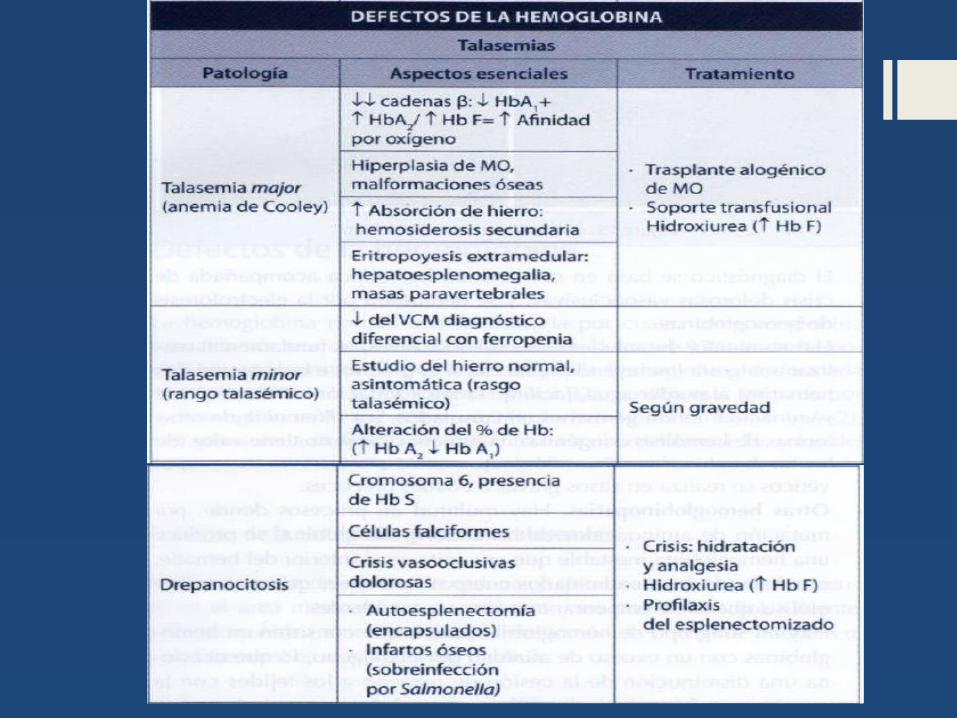
Talasemias
Talasemia mayor (anemia de cooley) o talasemia homocigota
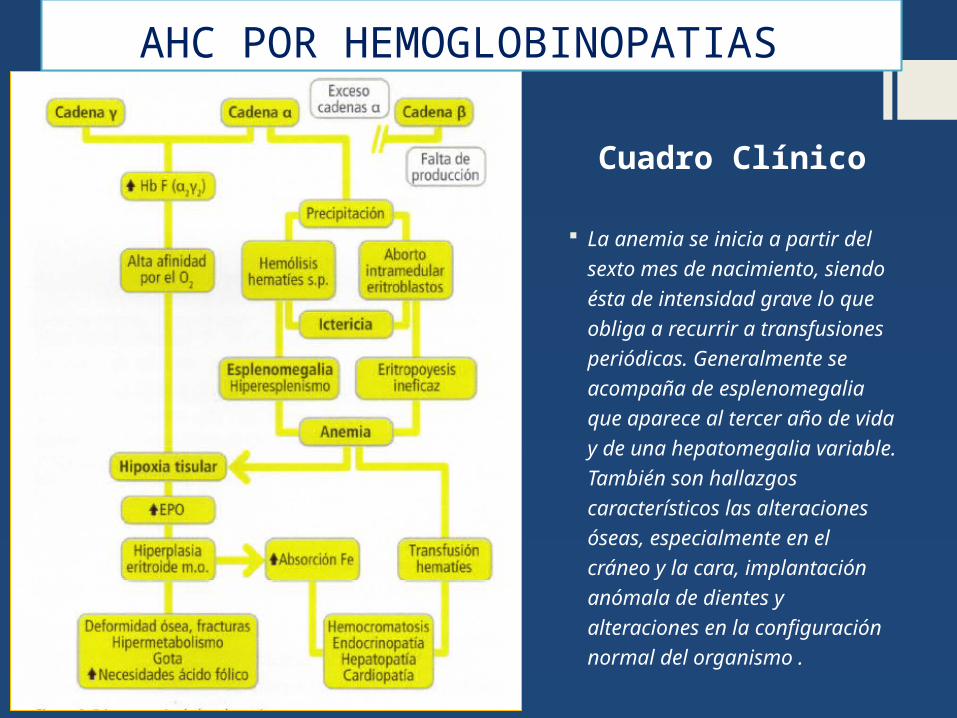
AHC POR HEMOGLOBINOPATIAS
La anemia se inicia a partir del sexto mes de nacimiento, siendo ésta de intensidad grave lo que obliga a recurrir a transfusiones periódicas. Generalmente se acompaña de esplenomegalia que aparece al tercer año de vida y de una hepatomegalia variable. También son hallazgos característicos las alteraciones óseas, especialmente en el cráneo y la cara, implantación anómala de dientes y alteraciones en la configuración normal del organismo .
Cuadro Clínico

AHC POR HEMOGLOBINOPATIAS
Mielograma: Los depósitos de hierro se encuentran incrementados y la celularidad hematopoyética se caracteriza por ser hiperplásica sobretodo en la serie eitroide.

Tratamiento
AHC POR HEMOGLOBINOPATIAS
Transfusiones sanguíneas: Tienen como objetivo mantener los niveles de hemoglobina alrededor de 10,0 g/ dL para asegurar un desarrollo psico motriz normal y una mejor calidad de vida del paciente.
Quelantes de Hierro: La deferoxamina es el más usado y menos tóxico. Este fármaco se administra por vía subcutánea a dosis de 50 a 60 mg/ Kg.Esplenectomía: Se recomienda en los pacientes con intensa esplenomegalia sobretodo si hay compresión de órganos vecinos e hiperesplenismo.
Transplante de Médula Ósea: siempre que sea posible, debe ser el trasplante alogénico de precursores hematopoyéticos, ya que se trata de una enfermedad genética.

AHC POR HEMOGLOBINOPATIAS
Corresponde a los estados heterocigotos para los genes de la cadena beta, en donde una de ellas se encuentra alterada mientras que la otra, es normal ( +/ y °/ ).
El cuadro clínico se caracteriza por la presencia de anemia muy leve y en muy pocos casos se puede presentar esplenomegalia.
DIAGNÓSTICO
• Hemograma: anemia leve de tipo microcítica hipocrómica con reticulocitosis. En el frotis periférico se puede encontrar la presencia de dianocitos y punteado basófilo.
• Médula ósea: aumento de la hemosiderina y maduración megaloblástica por consumo de folato
• Electroforesis de Hemoglobina: Característicamente se presenta un incremento de la hemoglobina A2, mientras que la hemoglobina fetal puede estar normal o discretamente elevada.
Se pueden observar glóbulos rojos sanguíneos de varias formas (poiquilocitosis), pálidos (hipocrómicos) y pequeños
(microcíticos),
Talasemia minor o rasgo talasémico (heterocigotos simples)

AHC POR HEMOGLOBINOPATIAS
TRATAMIENTOSu objetivo es básicamente preventivo y se
consigue mediante la administración de ácido fólico a dosis de 1 mg por día. No se debe
administrar suplementos de hierro por el riesgo de hemocromatosis .