
CONCENTRACION DE HIERRO ORGÁNICO EN …14:52Z-439… · Las hemoglobinopatías son alteraciones...
87
REPUBLICA BOLIVARIANA DE VENEZUELA LA UNIVERSIDAD DEL ZULIA FACULTAD DE MEDICINA DIVISIÓN DE ESTUDIOS PARA GRADUADOS POSTGRADO DE HEMATOLOGÍA HOSPITAL CENTRAL “Dr. URQUINAONA” CONCENTRACION DE HIERRO ORGÁNICO EN ANEMIAS HEREDITARIAS COMO PREVENCION DE HEMOSIDEROSIS. Trabajo Especial de Grado presentado ante la División de Estudios Para Graduados de la Facultad de Medicina, para optar al título de Especialista en Hematología. TUTOR ACADEMICO AUTOR Dr. Jorge Herrera Cepeda. M.C María Eugenia Vizcaíno. TUTOR METODOLOGICO Dra. Mery Guerra. MARACAIBO, MAYO 2011
-
Upload
truongtuyen -
Category
Documents
-
view
215 -
download
0
Transcript of CONCENTRACION DE HIERRO ORGÁNICO EN …14:52Z-439… · Las hemoglobinopatías son alteraciones...

REPUBLICA BOLIVARIANA DE VENEZUELA LA UNIVERSIDAD DEL ZULIA
FACULTAD DE MEDICINA DIVISIÓN DE ESTUDIOS PARA GRADUADOS
POSTGRADO DE HEMATOLOGÍA HOSPITAL CENTRAL “Dr. URQUINAONA”
CONCENTRACION DE HIERRO ORGÁNICO EN ANEMIAS
HEREDITARIAS COMO PREVENCION DE HEMOSIDEROSIS.
Trabajo Especial de Grado presentado ante la División de Estudios Para Graduados de la Facultad de Medicina, para optar al título de
Especialista en Hematología.
TUTOR ACADEMICO AUTOR Dr. Jorge Herrera Cepeda. M.C María Eugenia Vizcaíno. TUTOR METODOLOGICO Dra. Mery Guerra.
MARACAIBO, MAYO 2011

CONCENTRACION DE HIERRO ORGÁNICO EN ANEMIAS HEREDITARIAS COMO PREVENCION DE HEMOSIDEROSIS.

DEDICATORIA
A Dios por abrirme puertas, brindarme oportunidades y por su infinito amor. A mis padres por el apoyo incondicional necesario para superar cada una de las metas que me he propuesto, y estar siempre a mi lado. A mi esposo por su amor y apoyo. A mis hijos por ser mi motivo, iluminando mis días con su existencia y su dulzura.

AGRADECIMIENTOS A Dios por ayudarme en todo momento durante el largo camino de la vida. A mi familia por apoyarme e impulsarme para seguir adelante. A la Dra. Mery Guerra por sus enseñanzas sobre Metodología de la Investigación, responsabilidad, cumplimiento y sobre todo por su paciencia. A la Dra. Melvis de Vizcaíno, mi mami, digna de admiración, mi inspiración y modelo a seguir, sin ti esto no sería posible, simplemente GRACIAS!. Al Dr. Jorge Herrera por ser el maestro guía en esta etapa de mi formación. Al personal que labora en el servicio de Hematologia del hospital Dr Urquinaona, por su apoyo incondicional. A todos y cada uno de los adjuntos del servicio de Hematología del Hospital Central Dr. Urquinaona, por sus apreciadas orientaciones y por aclarar el rumbo a seguir en esta etapa. A todos los pacientes que tuve la oportunidad de tratar a través de la practica profesional, por el amor y la confianza que me brindaron. A todos mis compañeros de postgrado con los cuales compartí muchos momentos inolvidables. A todos y cada uno de los que me brindaron su apoyo…. Gracias.

V E R E D I C T O
Este jurado aprueba el trabajo especial de grado titulado: “CONCENTRACION DE HIERRO ORGÁNICO EN ANEMIAS HEREDITARIAS COMO PREVENCION DE HEMOSIDEROSIS”, elaborado por la Medica Cirujana María Eugenia Vizcaíno Arteaga, C.I. No. 14.370.828, presenta ante el Consejo Técnico de la División de Estudios para Graduados de la Facultad de Medicina en cumplimiento con el Artículo 45, Parágrafo 45.2 de la Sección Primera del reglamento de Estudios para Graduados de la Universidad del Zulia, como requisito para optar al Grado Académico de ESPECIALISTA EN HEMATOLOGIA. _____________________ Coordinador del Jurado Nombre y Apellidos. C.I. _____________________ Jurado Nombre y Apellidos. C.I. _____________________ Jurado Nombre y Apellidos. C.I.
MARACAIBO, Mayo 2011.

TABLA DE CONTENIDO
DEDICATORIA
Página
AGRADECIMIENTO
ABSTRACT RESUMEN TABLA DE CONTENIDO LISTA DE TABLAS INTRODUCCIÓN ...................................................................................... 14 CAPÍTULO I
Planteamiento del problema ........................................................... 17
Formulación del problema .............................................................. 21
Objetivos de la investigación .......................................................... 21
Justificación e importancia de la investigación ............................... 22
Delimitación de la investigación ..................................................... 24
Factibilidad y viabilidad de la investigación .................................... 24 CAPÍTULO II

Marco teórico conceptual ............................................................... 26
Antecedentes de la investigación ................................................... 26
Bases teóricas ............................................................................... 30
Marco teórico operacional .............................................................. 63
Definición conceptual y operacional ............................................... 63
Operacionalización de las variables ...............................................
64 CAPÍTULO III
Tipo de investigación ..................................................................... 66
Diseño de la investigación ............................................................. 66
Materiales y métodos ..................................................................... 67
Población ....................................................................................... 67
Muestra .......................................................................................... 67
Criterios de exclusión e inclusión ................................................... 67
Método........................................................................................... 67
Recolección de datos ..................................................................... 68
Análisis de datos ........................................................................... 68 CAPÍTULO IV
Resultados ..................................................................................... 70
Discusión ....................................................................................... 75 CAPÍTULO V

Conclusiones .................................................................................. 81
Recomendaciones .......................................................................... 82 LITERATURA CITADA .............................................................................. 83

INDICE DE TABLAS TABLA I
Características generales de los pacientes con Anemia Falciforme y Talasemia
estudiados………………………………………………………………………………….71
TABLA II
Distribución de los pacientes con Anemia Falciforme y Talasemia, según los
parámetros de hierro analizados…………………………………………………………72
TABLA III
Análisis estadístico de los parámetros de hierro medidos En los pacientes con Anemia
Falciforme y Talasemias estudiados………………………………..……………………73
TABLA IV
Correlación entre % de saturación de la transferrina y ferritina en los pacientes con
anemia falciforme y talasemia……………………………………………………………74.

13 Vizcaíno A., María E. (Autor), Herrera Cepeda Jorge (Tutor Académico); Guerra Velásquez, Mery (tutor Metodológico). “CONCENTRACION DE HIERRO ORGÁNICO EN PACIENTES CON ANEMIAS HEREDITARIA COMO PREVENCION DE HEMOSIDEROSIS”. (2011). Proyecto de Investigación. División de Estudios para Graduados. Facultad de Medicina. Universidad del Zulia.
RESUMEN
Objetivo: Determinar la concentración sérica de hierro orgánico como pronóstico de hemosiderosis, en pacientes con Anemia Falciforme (AF) y Talasemia atendidos en el Servicio de Hematología del Hospital Central Dr. Urquinaona y el Instituto Hematológico de Occidente Banco de Sangre del estado Zulia. Se realizó una investigación de tipo correlacional, transversal con un diseño no experimental y una muestra no probabilística de 26 pacientes. Material y Método: En ayunas se extrajo sangre venosa para determinar hierro sérico (Fe), % de Saturación de la Transferrina (%ST) y Ferritina. Resultados: la edad promedio fue 38.08 ± 12,96 años, predominó el sexo femenino (53,85%), el 69,24% correspondió a AF y 30,7% Talasemia. El promedio de transfusiones anual fue 11,92 ± 8,22. El 44,4% de AF y 50% de Talasemia presentaron un %ST por encima de su valor normal pero menor de 45% y ferritina por encima de su valor normal pero menor de 1000 ng/ml. El 22% de AF y 50% de Talasemia presentaron Hemocromatosis. En todos los pacientes con AHH el %ST fue 53,36±27,16 %, Ferritina: 1.608,67±1721,7 ng/ml y el número de transfusiones fue 11,92±8,38. Se encontró correlación significativa (p <0.01) ente el %ST y Ferritina y correlación significativa (p< 0.001) entre la edad y el número de transfusiones (p<0.01). El valor promedio y error estándar de Ferritina en AF fue 1.926±447,6 ng/ml, para Talasemia 889,15±557,16. El 50% de las Talasemias recibían tratamiento quelante de Fe y el 111,1% de AF. Conclusiones: la determinación de los parámetros de Fe en los pacientes con AF y Talasemia permite diagnosticar sobrecarga de Fe, debiéndose indicar tratamiento con quelantes de Hierro, y evaluación del tejido hepático y cardíaco, vigilando con regularidad la sobrecarga de Fe e identificar a tiempo efectos indeseables.
Palabras Clave: Hierro orgánico, Anemias Hemolíticas Hereditarias, Hemocromatosis.

14 Vizcaíno A., María E. (Autor), Herrera Cepeda Jorge (Tutor Académico); Guerra Velásquez, Mery (tutor Metodológico), "ORGANIC IRON CONCENTRATION IN PATIENTS WITH HEREDITARY ANEMIAS AS PREVENTION OF HEMOSIDEROSIS". (2011). Research project. For graduate studies Division. Faculty of medicine. University of Zulia.
ABSTRACT Objective: Determining the serum concentration of organic iron as prognosis of hemosiderosis in patients with sickle cell Anemia (AF) and Thalassemia in the service of Hematology of the Hospital Central Dr. Urquinaona and the hematological Institute of blood bank West of the State of Zulia. It carried out an investigation of correlational, transversal type with a non-experimental design and a sample not probability of 26 patients. Materials and methods: in fasting was extracted venous blood to determine serum iron (Fe), % of ferritin and transferrin saturation (% ST). Results: the mean age was 38.08 ± 12,96 years, dominated the female sex (53,85%), the % 69,24 corresponded to AF and 30.7% Thalassemia. The annual average of transfusion was 11,92 ± groaning. 44.4% Of AF and 50% of Thalassemia presented a ST % above its normal value but less than 45% and ferritin above its normal value but less than 1000 ng/ml. 22% Of AF and 50% of Thalassemia presented hemochromatosis. In all patients with AHH % ST was 53, 36±27, 16%, ferritin: 1.608, 67±1721, 7 ng/ml and the number of transfusions was 11, 92±8, 38. We found significant correlation (p < 0.01) between the ST % and ferritin and significant correlation between age and the number of transfusions (p < 0.001) (p < 0.01). The average value and error standard of ferritin in AF was 1. 926±447, 6 ng/ml, for thalassemia 889, 15±557, 16. 50% Of the Thalassemias received treatment chelator of faith and 111,1 per cent of AF. Conclusions: the determination of the parameters of organic iron in patients with AF and Thalassemia can diagnose overload of iron, should be treated with iron chelators, and evaluation of liver and heart tissue binders, regularly to monitor the overhead of iron and identify time undesirable effects. Keywords: organic iron, hereditary hemolytic anemias, hemochromatosis.

15
INTRODUCCION.

16
Las hemoglobinopatías son alteraciones cualitativas o cuantitativas de la globina,
secundarias a mutaciones genéticas, cuya consecuencia puede ser una modificación
estructural (hemoglobinopatías estructurales) o una disminución de la síntesis de una
cadena globínica estructuralmente normal (talasemias). Son el resultado de mutaciones
al nivel de alguno de los genes que codifican la síntesis de una determinada cadena
globínica: α, β, γ, ψ, y δ. Se consideran hemoglobinopatías solo aquellas mutaciones
que afectan regiones esenciales de la molécula y que, por tanto, poseen expresividad
clínica. En general, las mutaciones de aminoácidos situadas en la superficie de la
molécula solo producen modificaciones de la carga eléctrica, mientras que los
aminoácidos internos ocasionan, casi siempre, una importante alteración estructural y
funcional de la hemoglobina y su repercusión clínica suele ser mayor: anemia
hemolítica (hemoglobinas inestables), poliglobulia (hemoglobinas con alteración de su
afinidad por el oxígeno) o cianosis (hemoglobinas M).
Dos son las hemoglobinopatías que requieren un tratamiento regular: la talasemia
mayor y la drepanocitosis. El tratamiento es esencialmente paliativo y consiste en la
práctica de transfusiones periódicas, acompañadas de la administración de quelantes
del hierro y a veces de esplenectomía. Hoy en día el único tratamiento curativo es el
TMO alogénico, y está en fase experimental la manipulación genética.
Como contrapartida, el intenso régimen transfusional supone siempre un mayor
aporte de hierro al organismo y, por tanto, el peligro de una hemocromatosis que, por la
gravedad de sus complicaciones, puede ocasionar el fallecimiento del paciente casi
siempre antes de los 25 años de edad. Por ello, cualquier régimen de transfusiones se
acompaña de la administración de quelantes del hierro. Este gran aporte de hierro que
ocasiona daño orgánico, aunque se le reconoce que es un elemento imprescindible
para la salud, su exceso puede entrañar riesgos importantes.
Es por esto que el presente estudio se evalúan los niveles de hierro orgánico en
los pacientes con diagnostico de anemias hemolíticas hereditarias como prevención de
hemosiderosis, principal causa de morbimortalidad en estos pacientes.
La presente investigación se realizó en 5 capítulos: Capítulo I, análisis de la
situación objeto de estudio; Capítulo II, marco teórico; Capítulo III, marco metodológico;
Capítulo IV, resultados y discusión; y Capítulo V, conclusiones y recomendaciones.

17
CAPITULO I
EL PROBLEMA

18 PLANTEAMIENTO DEL PROBLEMA
Las Anemias Hemolíticas Hereditarias, como la Anemia Falciforme (AF)
Homocigota y la Talasemia, se caracterizan por tener una sobrevida acortada del
glóbulo rojo (GR) ocasionando cifras bajas de hemoglobina (Hb). Su frecuencia es alta
en el mundo, con una importante morbimortalidad (Bunn y col, 1986).
La AF llamada también drepanocítica, se caracteriza por presentar en su patrón
electroforético la presencia de la Hemoglobina S (HbS) en la cual existe una sustitución
de aminoácidos, el acido glutámico por valina (Williams, 2005). Este patrón se puede
detectar en el humano a los pocos meses de nacer, específicamente cuando ocurre el
reemplazo de la Hb Fetal que predomina durante los primeros meses de vida. La
sustitución de estos aminoácidos origina que la molécula de la hemoglobina se
cristalice produciendo la deformación de los GR que se tornan rígidos adquiriendo la
forma de hoz o falciforme, lo que entorpece su tránsito por los capilares pequeños. Este
proceso ocurre cuando desciende la PO2; es un círculo vicioso porque los eritrocitos
falciformes incrementan el estancamiento en los vasos sanguíneos generando un
mayor descenso de la PO2 acentuándose la falciformación (Bunn y col, 1986).
De igual manera, la Talasemia es un enfermedad congénita donde existe déficit en
la síntesis de una o más cadenas de globina (alfa o beta) de la Hb, produciéndose
acumulación intracelular de la cadena en exceso, así habrá α-Talasemia (exceso
cadenas de globina beta) y β-Talasemia (exceso cadenas de globina de alfa). En
ambos casos, se forman precipitados intracelulares en los eritroblastos que se
destruyen en la médula ósea y provocan eritropoyesis ineficaz. Los GR circulantes
también presentan precipitados en su interior por lo que son destruidos ocasionando
hemólisis. La más frecuente de las Talasemias es la β-Talasemia en la cual se
distinguen 3 grupos, a saber:
• Mayor o enfermedad de Cooley en esta se presenta una mayor clínica
• Menor en donde la clínica es poco manifiesta o ausente
• Intermedia presentándose de diferente grado clínico.

19
La Talasemia menor es la más frecuente y el diagnóstico casi siempre es casual;
se caracteriza por anemia discreta o inexistente e hipocromía, por ello fácilmente se
trata como una ferropenia, sometiendo al enfermo a una nociva sobrecarga de hierro
(Weatherall y col, 1991).
En cuanto a las frecuencias de estas enfermedades, la AF alcanza su mayor
frecuencia en el centro de África y la Talasemia mayor en el Mediterráneo donde la alfa
talasemia es la más frecuente en el Medio Oriente (Turgeon y col, 2006). En Venezuela
se ha encontrado una frecuencia de 9% para las hemoglobinopatías, siendo la la HbS
más frecuente con un 69,8%. Se evidenció también un 2% para la beta talasemia
asociada con HbS, por lo que se le reconoce como un problema de salud pública
(Arends y col, 2007). Este hecho es particularmente importante de conocer pues se
reporta que el porcentaje de HbS oscila entre 0-7% dependiendo del componente
africano de la región (Salazar-Lugo, 2004); en estado Zulia, específicamente en Isla de
Toas y sus regiones vecinas, se caracterizan por tener una población con
características fenotípicas caucásicas, por lo que es quizás el estado de Venezuela que
presenta una mayor frecuencia, así se describe un 13% en Isla de Toas (Pineda-Del
Villar y Borjas M, 1986) y 6,4% en las poblaciones vecinas (Torres-Guerra y col, 1993).
Tanto en la AF como en la Talasemia, se hace necesaria la administración de
transfusiones de eritrocitos desde tempranas edades de la vida, con el riesgo que estas
conllevan como la sobrecarga de hierro (Fe), entre otras. Esta sobrecarga de Fe agrava
la ya existente, producto de la destrucción crónica de los eritrocitos con la consecuente
liberación del Fe que se deposita en los diferentes tejidos (Gabutti y col, 1994). En
ambos casos, existe una oferta de Fe mayor de la que el organismo puede procesar y
al no existir un mecanismo excretor, regulador y corrector de la cuantía de ese hierro
corporal, se va acumulando progresivamente en las células parenquimatosas,
quedando excluidos hasta estadios muy avanzados, en las células del Sistema
Mononuclear Fagocítico, dentro del cual pertenecen las células de Kuppfer (Parkkila y
col, 2000).
La sobrecarga de Fe es mayor en pacientes con Talasemia, pues se le añade la
eritropoyesis ineficaz que las caracteriza, conduciendo a un aumento de la absorción de

20 Fe, lo que implica una situación paradójica pues existe anemia y además sobrecarga de
Fe (Papanikolaou y col, 2005).
Paralelamente se describe en las AF y las Talasemias, una importante frecuencia
de enfermedades hepáticas crónicas como la hepatitis por virus C, B, esteatohepatitis
no alcohólica, etc., producto principalmente de las transfusiones; en estas
enfermedades se produce acumulación de hierro, por lo que la sobrecarga de Fe podría
ser mayor en estos pacientes (Powell y col, 1994).
Ahora bien, todos los esfuerzos que se han hecho para aumentar la sobrevida de
los pacientes con AF y Talasemia, tanto en cantidad como en calidad, están dirigidos
principalmente a la terapia transfusional, que es un tratamiento esencialmente paliativo,
pues el tratamiento curativo es la manipulación genética, que en la actualidad se
encuentra en fase experimental, disponiéndose solo del trasplante de médula ósea
alogénica. En Venezuela, esta última terapia se realiza con poca frecuencia, por la
dificultad que tienen los pacientes para acceder a los centros públicos que lo realizan,
pues la demanda es mayor que la oferta y en los centros privados el costo es elevado.
En este sentido, se ha diseñado un programa de terapia transfusional a través de
(Malcorra y col, 2001):
• Un régimen clásico o transfusión a libre demanda, en donde solo se transfunde
cuando la hemoglobina desciende por debajo de <70-80 g/L
• El régimen de hipertransfusión que consiste en administrar tantas transfusiones
como sean necesarias para evitar que la hemoglobina descienda por debajo de 100 g/L,
evitándose la disminución de la eritropoyesis y con ello una drástica disminución de la
absorción de hierro intestinal.
• También se describe una variante de la hipertransfusión llamada
supertransfusión, para mantener un hematocrito por encima del 35% (Hb > 120 g/L).
Estos dos últimos regímenes requieren transfundir al paciente de 2 a 3
concentrados eritrocitarios cada 2 a 4 semanas. Las ventajas que tiene el régimen de
hipertransfusión es que se produce un mejor desarrollo óseo, menor esplenomegalia y
mejor calidad de vida, pero tiene el inconveniente de representar una mayor sobrecarga

21 de hierro o hemocromatosis, entre otros efectos adverso de la transfusión (Malcorra y
col, 2001).
Por lo antes señalado, es necesario contrarrestar el efecto de sobrecarga de Fe
que las numerosas transfusiones de GR ocasionan a los pacientes con anemias
hemolíticas hereditarias, ya que tienen el riesgo de producir hemocromatosis, cuyas
complicaciones son graves y responsables del fallecimiento del paciente antes de los
25 años de edad. Por ello, cualquier régimen transfusional debe acompañarse de la
administración de quelantes del hierro, entre ellos, el menos tóxico es el Mesilato de
Deferroxiamina (Desferal, DFO), pero tiene el inconveniente de tener un alto costo y se
amerita conocer la ferrocinetica del paciente antes y después de la administración del
mismo (Kwiatkowski y col, 2004).
Al respecto, en Venezuela se contempla la dotación de Deferasirox de forma
gratuita en pacientes que requieren múltiples transfusiones de GR, a través de Instituto
Venezolano de los Seguros Sociales (IVSS); sin embargo, en el estado Zulia, esta
modalidad terapéutica es subutilizada, ya que los pacientes, muchas veces no siguen
las indicaciones médicas o porque se desconoce sobre el mismo (Aparicio y col, 2007).
El diagnóstico de sobrecarga de hierro se basa en la sospecha clínica y la
determinación de los parámetros bioquímicos como el hierro sérico o sideremia, que no
tiene valor por sí solo, pero es necesario para conocer el porcentaje de saturación de la
transferrina (%ST), el cual corresponde al cociente entre el hierro sérico y la capacidad
total de fijación del hierro con la transferrina (CTFH). Este valor aislado puede ser
normal en las mujeres menores de 30 años con Hemocromatosis Hereditaria (HH) o dar
resultados falsos positivos si no se realiza en ayunas, y está influido además por las
variaciones circadianas y enfermedades inflamatorias. El %ST se eleva más
precozmente que la ferritina plasmática y se ha descrito que un valor mayor de 45%
debe poner en marcha el estudio diagnóstico, especialmente cuando es mayor del 50%
en mujeres y 60% en varones (Powell y col, 2004; Bacon y col, 1996; Ladero y col,
2005; Roa y col, 2001).

22
En la ciudad de Maracaibo, la mayoría de los pacientes con Anemia Falciforme y
Talasemia son atendidos en el Servicio de Hematología del Hospital Central, el Instituto
Hematológico de Occidente-Banco de Sangre del Estado Zulia y el Hospital
Universitario de Maracaibo, sedes hospitalarias donde se imparte además el Postgrado
Universitario de Hematología. En los dos primeros centros dispensadores de salud, se
realiza el diagnóstico, control y tratamiento de estas patologías. De allí que se plantea
realizar el presente estudio con el objetivo de analizar el estado del hierro orgánico en
pacientes con Anemia Falciforme y Talasemia, lo que permitirá determinar la población
con sobrecarga de Fe y recomendar los correctivos correspondientes.
1.1 FORMULACIÓN DEL PROBLEMA ¿Tienen los niveles de hierro orgánico valor pronostico de hemosiderosis en pacientes
con anemia hemolítica hereditaria?
1.2. OBJETIVOS DE LA INVESTIGACIÓN
1.2.1 Objetivo General: Determinar concentración sérica de hierro orgánico como pronostico de
hemosiderosis, en los pacientes Anemia Falciforme y Talasemia.
1.2.2 Objetivos Específicos:
• Caracterizar la población de AF y Talasemia estudiada, según la edad, sexo y
número de transfusiones por año.
• Determinar las concentraciones séricas de hierro sérico, TIBC, %ST, Ferritina sérica.
• Determinar el número de pacientes con AF y Talasemia hemosiderosis.
• Relacionar el %ST, Ferritina sérica con el numero de concentrados globular
administrado a los pacientes estudiados

23 1.3.-JUSTIFICACION
Teóricamente, la presente investigación se justifica, ya que analiza la
concentración de hierro orgánico en pacientes con anemia hemolítica hereditaria, para
determinar en diagnostico de hemosiderosis.
Las consecuencias clínicas que produce una sobrecarga de hierro pueden
prevenirse a través del diagnóstico temprano y la indicación de un tratamiento
adecuado, mucho más importante en pacientes que reciben transfusiones de eritrocitos
con frecuencia como aquellos con diagnóstico de anemia hemolítica.
En este sentido, en las anemias hemolíticas hereditarias se produce un incremento
de hierro en sangre y en tejidos por el aumento en la destrucción eritrocitaria que los
caracteriza. El aumento en la actividad de la médula ósea puede compensar de manera
temporal esta reducción, pero cuando la médula ósea no puede aumentar la producción
de eritrocitos para contrarrestar la pérdida de las células a causa de la hemólisis, se
produce anemia severa.
De todas las anemias hemolíticas hereditarias, la AF y la Talasemia son las más
frecuentes. La AF alcanza su mayor prevalencia en el centro de África, mientras que la
Talasemia mayor puede rastrearse hasta el Mediterráneo. En el Medio Oriente la
hemoglobinopatía más frecuente es la alfa talasemia (Turgeon y col, 2006). En
Venezuela se encontro una elevada frecuencia de hemoglobinopatías (9%), siendo la
HbS la variante más encontrada (69,8%), también observaron beta-talasemia asociada
con HbS (2%), constituyéndose en un problema de salud pública (Arends y col, 2007).
Por su parte, Salazar Lugo reporta un porcentaje de HbS que oscila entre 0-7%
dependiendo del componente africano de la región (Salazar-Lugo, 2004), así, en la
región zuliana específicamente en la Isla de Toas, la población tiene características
fenotípicas caucasoides pero tiene una frecuencia del 13% (30), mientras que en las
poblaciones vecinas que tienen las mismas características fenotípicas, el porcentaje del
gen es de 6,4% (Torres-Guerra Ey col, 1993), como se observa en el rango más alto
indicado por Salazar.

24
Lo antes descrito nos hace suponer que el riego de ocurrencia de enfermedad
transfusional como la sobrecarga de Fe, es mayor en los pacientes con AF y Talasemia
en el estado Zulia, siendo los casos con anemia grave quienes necesitan un mayor
número de transfusiones para prevenir la muerte, con la consecuente acumulación de
Fe proveniente de la sangre transfundida y de la hemolisis que sufren, tal como lo
describe Muller y colaboradores (Muller y col, 1993), sobre todo en aquellos pacientes
con predisposición genética a sufrir de hemocromatosis hereditaria, representando un
efecto tóxico acumulativo e inevitable para quienes reciben múltiples transfusiones
sanguíneas.
Al respecto, Jeng y colaboradores en California, estudiaron la asociación entre el
gen de la hemocromatosis hereditaria y la AF, reportando 2,3% de pacientes la
mutación para heterocigotos y 6,8% en heterocigotos (Jeng y col, 2003). En nuestro
país un estudio similar no ha sido reportado, lo que supone un mayor riesgo para estos
pacientes si no se indica dentro de su protocolo de tratamiento, agentes quelantes del
hierro.
Científicamente se justifica ya que permite, profundizar enfoques teóricos de
expertos en AF y la Talasemia, lo que amplía el conocimiento existente en cuanto a su,
epidemiología, fisiopatogenia, etiopatogenia que interviene en su aparición, así como en
la terapia transfusional y sus consecuencias. Todos estos aspectos permiten la
actualización durante la última década especialmente sobre los diversos criterios
referentes al tratamiento.
Por otra parte, desde el punto de vista práctico, el estudio se justifica debido al
análisis del estado del hierro orgánico en pacientes con AF y Talasemia, que permitirá
determinar el porcentaje de pacientes con sobrecarga de Fe y las necesidades del
tratamiento quelantes, de tal manera que pueda contribuir a prevenir la hemosiderosis,
sus complicaciones y realizar tempranas modificaciones en el tratamiento.
En su carácter metodológico resulta de suma importancia, porque a través de la
utilización de procedimientos diagnóstico comunes que se realizan en los laboratorios
de los hospitales y accesibles para los pacientes, se podrá diagnosticar la sobrecarga

25 de FE, con lo cual se pretende ofrecer una metodología sencilla con beneficio para la
comunidad en general, además aporta un instrumento que clasifica el pronóstico de los
pacientes con las patologías antes mencionadas, el cual podrá ser aplicado en otras
organizaciones del ramo.
1.4 DELIMITACION DE LA INVESTIGACION Este estudio se enmarca dentro de las líneas de investigación del Servicio de
Hematología del Hospital Central Dr. Urquinaona y el Instituto Hematológico de
Occidente Banco de Sangre del estado Zulia. Se llevara a cabo en el Servicio de
Hematología del Hospital Central Dr. Urquinaona, ubicado en la Avenida 2 El Milagro de
la Ciudad de Maracaibo, Estado Zulia, y el Instituto Hematológico de Occidente Banco
de Sangre del estado Zulia, ubicado en el sector veritas, de Maracaibo. En un periodo
comprendido desde abril 2009 hasta julio del 2010 con una duración de 15 meses.
1.5. FACTIBILIDAD Y VIABILIDAD El presente trabajo es factible hacerlo ya que se dispone de los pacientes
diagnosticados con AF y Talasemia que acuden al Servicio de Hematología del
Hospital Central Dr. Urquinaona y el Instituto Hematológico de Occidente Banco de
Sangre del estado Zulia, además se tiene la posibilidad de conseguir la ayuda
económica para obtener el Kit de reactivos necesario para la determinación de hierro
orgánico sérico.
26
CAPITULO II
MARCO TEORICO.

27
MARCO TEÓRICO CONCEPTUAL 2. Antecedentes de la Investigación
Toda investigación que se desarrollará requiere un severo proceso metodológico
para permitir paso a paso el alcance de los objetivos, el cual requiere como condición
la revisión de antecedentes, los cuales permiten reforzar los resultados obtenidos en el
estudio. En aras de cumplir con este fin se presentan algunos de los trabajos de
investigación previos que abordan la variable de estudio y se consideran pertinentes
señalarlos, considerándose como aportes a esta investigación.
En ese sentido, se encuentra el estudio realizado por Vázquez y col (2002),
quienes realizaron una revisión sistemática de las Reacciones post-transfusionales, el
objetivo fue identificar y prevenir precozmente las complicaciones de las transfusiones
de hemoderivados alogénicos. En la misma ellos concluyeron que la sobrecarga de
hierro inducida por transfusiones es una consecuencia fatal frecuente con la transfusión
crónica para ciertos tipos de anemias. Los niños con talasemia constituyen el grupo
aislado más grande afectado. Cada mililitro de eritrocitos deposita 1,08 mg de hierro en
los tejidos a medida que dichos eritrocitos envejecen y mueren. El depósito de hierro
comienza a afectar las funciones endocrinas, hepática y cardíaca cuando la carga
alcanza los 20 g, el equivalente a 100 unidades de concentrado de globulos rojos
(CGR). Las complicaciones cardíacas letales ocurren con 60 g (300 unidades CGR).
Por lo tanto, debe considerarse la terapia con quelantes de hierro en todos los
pacientes que requieran de transfusiones crónicas.
Koren y col (2009), llevaron a cabo una investigación denominada hierro no unido
a Transferrina (NTBI) y sobrecarga de hierro en anemia falciforme: estudio comparativo
entre pacientes con AF y β-Talasemia. Cuyo objetivo fue estudiar la situación del hierro
orgánico, incluido NTBI, el efecto de la sobrecarga de hierro, y comparar los resultados
entre los pacientes con AF y β-Talasemia. Se utilizó un método prospectivo y fueron
inscritos 36 pacientes con diagnostico de AF y 43 con Talasemia, se realizaron
estudios de hierro sérico en ambos grupos. Los resultados indicaron que ninguno de los
pacientes con AF tenía síntomas clínicos de sobrecarga hierro, Sólo el 5% de los

28 pacientes con AF presentaron valores NTBI en la zona gris (0,4 unidades) y ninguno
tuvo valores positivos. Por el contrario, 32% de los pacientes con talasemia mayor y 7%
con talasemia Intermedia tuvieron valores NTBI por encima de 0,6, nivel que está en el
rango patológico positivo. Asimismo, 9% de pacientes con talasemia, pero sólo el 2%
con AF tenía niveles positivos de hierro sérico (HS). Llegándose a la conclusión de que
los parámetros de estado del hierro en los pacientes con AF, incluso después de recibir
transfusiones frecuentes, son diferentes en comparación con los pacientes con
talasemia. Los bajos niveles NTBI y HS encontrados pacientes con AF están en
consonancia con la ausencia de signos clínicos de la sobrecarga de hierro en esta
enfermedad.
Por su parte, Brown y col (2009), efectuaron una investigación titulada
Sobrecarga de hierro hepática en niños con anemia drepanocítica en terapia de
transfusión crónica. Siendo el objetivo del mismo explorar las relaciones entre las
variables contenido de hierro hepática (HIC), edad y número de transfusiones, en una
cohorte de niños con AF en terapia de transfusión crónica, y dosis bajas de quelantes
de hierro. El mismo se basó en un estudio prospectivo, en el cual se incluyeron 27
niños con AF, se realizaron biopsias Hepática antes de iniciar la terapia de quelación,
fueron evaluadas las terapias de transfusión crónica para anotar el resultado histológico
y determinación de HIC, asimismo, se determino valores de saturación de ferritina y
hierro sérico durante 6 meses antes de la toma de biopsia. La duración y el volumen
total de transfusión se obtuvieron de los registros médicos. Todos los niños fueron
negativos para el virus de inmunodeficiencia humana, el virus de hepatitis b y las
infecciones de virus de la hepatitis C.
La edad promedio en la biopsia fue 10.95 +/-3.34 años. La significancia del
volumen total y duración de las transfusiones fueron 17,4 +/-9.6 L y 50.0+/-26.6 meses,
respectivamente. Para el análisis estadístico se utilizo el coeficiente correlación de
Pearson dando como resultado correlaciones significativas entre HIC y valor de hierro
histológico, ferritina serica, saturación de hierro, edad y volumen de transfusión.
Después de ajustar por volumen de transfusión, se vio sólo una correlación significativa
entre volumen HIC y transfusión. Significancia de HIC peso seco mg/g 21.8 +/-10.4,
asimismo se observo fibrosis en 10 pacientes e inflamación lobular en 9. Por otra parte,

29 encontraron que HIC fue superior en biopsias con fibrosis (28,2 +/-3,8 mg/g) que en
biopsias sin fibrosis (17,6 +/-18,3 mg/g; P = 0,012). La HIC no difirieron entre biopsias
con inflamación lobular (25,5 +/-4.0 mg/g) y biopsias sin inflamación (19,9 +/-2,5 mg/g;
P = 0,22). Estos resultados muestran que el volumen de transfusión proporciona una
idea de cómo se encuentran los niveles de hierro hepático.
Asimismo, Rodolfo y col (2008) publicaron un estudio llamado “Registro de
Pacientes en América Latina con Hemosiderosis transfusional” (Registry of Latin
Americans with Transfusional Hemosiderosis -RELATH study). El objetivo de este
trabajo consistió en recopilar datos retrospectivos en pacientes con Hemosiderosis
transfusional (HT), en los siguientes países: Argentina, Brasil, Colombia, México,
Panamá, Perú y Venezuela. Los casos fueron agrupados por centros Hematologícos
con gran volumen de pacientes, que ofrecen atención hematologica terciaria en las
grandes ciudades a través de un instrumento metodológico diseñado para el estudio.
Se eligieron pacientes con > 2 años de edad, los cuales formaban parte de la consulta
en las instituciones participantes desde enero de 2004, y que presentaran cualquier
desorden que requiriera transfusiones crónicas de eritrocitos (RBC), recepción de > 9
unidades de RBC, al menos un valor de ferritina suero > 1000 mcg/L, o un contenido de
hierro de hígado (HIC) > 2 mg/g de peso seco (las leucemias fueron excluidos).
Entre Sep/06 y Ene/08, se recopilo información de 859 pacientes (pts), 850 de los
cuales son evaluables. La edad promedio fue de 29.2±20.1 (rango de 2 a 93), y el
53,2% de pts eran mujeres. Distribución étnica fue africano (37,4%) y de ascendencia
caucásica (31,4%), hispana (26,4%) y otros (4,8%). Los diagnósticos más frecuentes
fueron AF (48,9%), beta-talasemia importantes (15,4%), la anemia aplásica (9,5%) y
Síndrome mielodisplasico (7,6%, de los cuales 41,5% tenían anemia refractaria).
Transfusión de RBC fue > 9 en 100% y > 19 de 87,5% de pts (la edad era menor en
este último grupo, p = 0,002) y significancia determinación de ferritina de HIC de mcg/l.
2627±1964 no estaba disponible o no fue hecho en 89,5% de los casos; cuando se
realizo, el resultado fue elevada en 39,7%. El nivel de hemoglobina en la que se indicó
transfusión fue de 7 a 10 g/dL en un 67,7% y 6 g/dL o menos en 23,0% (N/A en 9,2%).

30
El número medio de las transfusiones que recibió fue 12.2±9.2/yr (rango de 1 a
80). La sobrecarga de hierro se evaluó mediante ferritina (90.0%), saturación
transferrina (22,1%) y ecocardiograma (21,3%). Las complicaciones de HT fueron
reportadas en 82,1% de los casos (62,5% de pts tuvo complicaciones hepáticas, 26,8%
endocrinas, cardíacas de 17,9%). La Terapia de quelación de hierro fue dado a 45,1%
de pts, con más frecuencia sobre la base de ferritina (84,5%), número de transfusiones
(29,9%) y complicación de sobrecarga de hierro (4,4%). Deferoxamina (88,9%) y
deferasirox (13,3%) fueron los quelantes utilizados con más frecuencia. En la mayoría
de los casos, el tratamiento estaba aún en curso, pero se reportaron los motivos de la
interrupción de tratamiento por cumplimiento deficiente (5,1%), drogas ya no está
disponible (3,4%) y negativa del pt (2,1%).
Se concluyo que este informe provisional del estudio RELATH muestra que un
registro es factible y puede proporcionar información valiosa sobre HT y patrones de
uso de la terapia de quelación del hierro en América Latina (AL). Además, el estudio
sugiere hasta ahora que la mayoría pacientes con transfusión crónica en AL
desarrollan HT, cuyas complicaciones podrían evitarse mediante el uso más eficaz de
quelantes de hierro.
León- González y colaboradores (2001), publicaron un estudio llamado
Hemocromatosis: Revisión literaria y presentación de casos clínicos, con el objetivo de
realizar una revisión profunda sobre el concepto de hemocromatosis, sus causas,
frecuencia, tratamientos y presentación de casos clínicos ilustrativos, en el mismo se
concluyo, que las principales causas de hemocromatosis secundarias son: Enfermedad
hepática, Talasemia mayor, Porfiria cutánea tardía, medicamentosa, Transfusional,
Anemia sideroblástica, Anemia hemolítica crónica, Atransferrinemia, nutricional.
Asimismo, se encontró que la hemocromatosis primaria (HFE) se considera el
trastorno genético más frecuente en la raza blanca (7%) con una prevalencia de 1 en
300 en los países sajones y europeos. Un hecho importante destacado en este estudio,
es la excelente respuesta al tratamiento con flebotomías y quelantes de hierro como la
desferroxamina, y que el inicio temprano del mismo previene complicaciones que
comprometen la vida del paciente, además de brindar una mejor calidad de vida.

31
Evans y col. (2003), en su trabajo titulado Talasemia asociada al embarazo.
Analizaron pacientes con talasemia embarazadas, y su objetivo fue estudiar el manejo
y las complicaciones de esta patología. Los resultados evidenciaron que es infrecuente
que la Talasemia mayor represente un problema para el obstetra, dado que la
sobrecarga de hierro asociada a esta patología suele provocar falta de crecimiento
puberal y retraso del desarrollo sexual. A menudo, estas pacientes son infértiles y
anovulatorias, y presentan hipogonadismo hipogonadotrófico por depósito de
hemosiderina en el hipotálamo y la hipófesis. Se han descrito alrededor de 14
embarazos en este grupo con cinco pérdidas fetales.
El embarazo puede precipitar una insuficiencia cardíaca; en este estado de
sobrecarga de hierro, estas pacientes requieren un monitoreo cardiovascular estricto
con mantenimiento de un nivel de hemoglobina de 10 g/dl. La talasemia intermedia y la
talasemia menor no constituyen impedimento para el embarazo, pero requieren
suplementos adicionales de ácido fólico. Sin embargo, la talasemia mayor como
demuestra la evolución de las pacientes estudiadas no sólo es peligrosa para el feto el
que generalmente muere, sino también para la madre que puede resultar con secuelas
severas.
2.2 Bases Teóricas: El Hematíe
El hematíe es una célula que presenta importantes diferencias con respecto a
otras células del organismo. En primer lugar, no tiene núcleo, por lo que no posee la
capacidad de división. Ni tampoco tiene mitocondrias o ribosomas, ni ADN o ARN. No
obtiene energía del ciclo de Krebs, y no tiene un sistema de transporte de electrones
para la fosforilación oxidativa. A pesar de estas deficiencias, el hematíe es una célula
compleja y metabólicamente activa cuya vida media es de alrededor de 120 días.
La integridad del hematíe depende de la interacción de tres unidades celulares,
que lo capacitan para realizar su función primaria de transporte de oxígeno y CO2.
Estas tres unidades celulares son la hemoglobina, la membrana eritrocitaria, y los

32 elementos solubles intracelulares (enzimas, coenzimas, y substratos del metabolismo
de la glucosa). La alteración de una de estas unidades celulares da lugar a alteraciones
en las otras dos, dando como resultado un acortamiento de la vida media eritrocitaria
(Malcorra JJ. 2001; Fairbanks VF, 1980).
HEMOGLOBINA
Cada molécula de hemoglobina (Hb) está formada por cuatro subunidades
protéicas denominadas globinas y 4 grupos hemo. Las subunidades proteicas al unirse
entre sí forman una estructura globular en la que se disponen unas cavidades donde se
alojan los grupos hemo. En su región central, las 4 cadenas delimitan un espacio para
el 2,3-difosfoglicerato (2,3-DPG) metabolito derivado de la glucolisis anaerobia que
favorece la liberación de oxígeno. El grupo hemo, sintetizado por los eritroblastos, es
una porfirina que posee un átomo de hierro en estado reducido, de las seis valencias de
coordinación que posee, una se une a la globina y otra se fija reversiblemente al
oxígeno. (Malcorra JJ. 2001; Fairbanks VF, 1980).
La unión del oxígeno al grupo hemo sólo es posible cuando el hierro se halla en
forma reducida (Fe++), y cuando se oxida (Fe+++), la hemoglobina se transforma en
matehemoglobina que no puede fijar el oxígeno, careciendo, por lo tanto, de función
respiratoria.
La naturaleza de las cadenas globínicas determina diferentes tipos de
hemoglobinas, siendo la llamada hemoglobina A (HbA) la predominante en el individuo
adulto normal.
La HbA constituye aproximadamente el 98% de la totalidad del contenido
hemoglobínico eritrocitario y está formada por dos cadenas α y dos cadenas β (α2β2)
que al unirse entre sí adoptan una configuración espacial globular, necesaria para el
desarrollo de la función respiratoria. El 2% restante está constituido por hemoglobina A2
(HbA2) formada por dos cadenas α y dos cadenas δ (α2δ2) y hemoglobina fetal (HbF)
formada por dos cadenas α y dos cadenas γ (α2γ2).

33
Durante el desarrollo embrionario y fetal existen cuatro hemoglobinas principales:
Hb Gower-1; Hb Gower-2; Hb Portland y Hb F. Después del 2° mes de gestación las
dos hemoglobinas Gower desaparecen en condiciones normales. La Hb Portland puede
prolongar su presencia hasta el nacimiento aunque en cantidades minúsculas. No así la
Hb F, que representa alrededor del 80% del contenido hemoglobínico de los hematíes
del recién nacido, correspondiendo el resto a Hb A.
El declive en la síntesis de Hb F es rápido en condiciones normales, de tal forma
que a los seis meses de vida sólo se detecta un 5% de esta hemoglobina en el niño. Sin
embargo, existen fluctuaciones importantes según los grupos étnicos. En lo que se
refiere a la Hb A, su síntesis comienza en edades tempranas de la vida fetal (segundo
mes) y su progresión es rápida una vez que se ha producido el parto.
La Hb A2, comienza a sintetizarse en el tercer trimestre del embarazo y está
presente en cantidades apenas perceptibles en el momento del nacimiento. Se puede
concluir que hacia la 40° semana de vida extrauterina, el niño presenta ya los
porcentajes hemoglobínicos propios del adulto. La cadena α consta de 141 aminoácidos
y las cadenas β, δ, ψ, y γ constan de 146 aminoácidos. Los genes α están localizados
en el cromosoma 16, y los β, γ, y δ en el cromosoma 11
Hemoglobinopatías (Malcorra JJ. 2001; Williams 2005)
Las hemoglobinopatías son alteraciones cualitativas o cuantitativas de la globina,
secundarias a mutaciones genéticas, cuya consecuencia puede ser una modificación
estructural (hemoglobinopatías estructurales) o una disminución de la síntesis de una
cadena globínica estructuralmente normal (talasemias).
Hemoglobinopatías estructurales
Son el resultado de mutaciones al nivel de alguno de los genes que codifican la
síntesis de una determinada cadena globínica: α, β, γ, ψ, y δ. Se consideran
hemoglobinopatías solo aquellas mutaciones que afectan regiones esenciales de la
molécula y que, por tanto, poseen expresividad clínica. En general, las mutaciones de

34 aminoácidos situadas en la superficie de la molécula solo producen modificaciones de
la carga eléctrica, mientras que los aminoácidos internos ocasionan, casi siempre, una
importante alteración estructural y funcional de la hemoglobina y su repercusión clínica
suele ser mayor: anemia hemolítica (hemoglobinas inestables), poliglobulia
(hemoglobinas con alteración de su afinidad por el oxígeno) o cianosis (hemoglobinas
M).
Clasificación Clínica
– Variantes por mutación superficial.
Síndromes Drepanocíticos:
a) Rasgo drepanocítico (AS)
b) Anemia drepanocítica (SS)
c) Dobles estados heterocigotos (SC)(SD), (S-ß-talasemia)
– Variantes de Hb inestable (anemia hemolítica congénita con cuerpos de Heinz).
– Variantes de Hb con elevada afinidad por el oxígeno (eritrocitosis familiar).
– Hemoglobinas M (cianosis familiar).
Los síndromes drepanocíticos sólo dan clínica en el estado homocigoto o doble
estado heterocigoto. Por el contrario, las variantes inestables, las de alta afinidad por el
oxígeno, y las hemoglobinas M solo se encuentran en estado heterocigoto.
Síndromes drepanocíticos (Malcorra JJ. 2001; Fairbanks VF, 1980)
Hemoglobina S
La Hb S tiene una alta prevalencia en África Tropical, en donde se observan
heterocigotos en el 20 y hasta el 40% de la población. La Hb S se puede encontrar en
tres formas diferentes como ya hemos visto antes. La Hb S se produce por la
sustitución del ácido glutámico por la valina. Al descender la PO2 la sustitución de dicho
aminoácido origina que la molécula de la hemoglobina cristalice, deformando los
hematíes, volviéndolos falciformes y rígidos, e impidiendo su tránsito por los capilares
pequeños. El proceso origina un círculo vicioso: los eritrocitos falciformes incrementan
el estancamiento, desciende mas la PO2 y se acentúa la falciformación. Si esto se
mantiene mucho tiempo, se lesiona la membrana celular, permitiendo el paso de calcio

35 al interior de la célula, lo que determina rigidez de la membrana. En estas condiciones
los hematíes son eliminados de la circulación por el SMF.
Hemoglobina AS o forma heterocigota (rasgo drepanocítico)
Los portadores de este trastorno son asintomáticos. Ocasionalmente sufren
hematurias e infartos esplénicos cuando se exponen a situaciones de hipoxia
prolongada (anestesia general y procesos neumónicos). La morfología eritrocitaria es
normal y no se observan drepanocitos en el frotis de sangre. Hay varias pruebas de
laboratorio para poner en evidencia la presencia de Hb S:
– El test de falciformación: se basa en la desoxigenación de la sangre in vitro cuando se
pone en contacto con un agente reductor.
– Prueba de solubilidad: consiste en la observación de que la hemoglobina S en estado
reducido, es muy insoluble en tampón fosfato concentrado.
– Electroforesis de Hb: Se verá una banda de desplazamiento lento con relación a la Hb
A. En los verdaderos heterocigotos la proporción de Hb S oscila entre un 35 y un
45% del total.
Hemoglobina S homocigota (SS) o anemia drepanocítica
La anemia drepanocítica o drepanocitosis comprende un grupo de anemias
hemolíticas crónicas hereditarias en las que está presente el gen de la hemoglobina
(Hb) S. Este gen está ampliamente difundido en África y fue trasladado a América
mediante el comercio de esclavos y en muchos de estos países se considera un
problema de salud pública. La frecuencia del estado de portador AS es de 3,08 % en la
población general. La anemia drepanocítica o hemoglobinopatía SS es la de mayor
incidencia; le siguen en orden de frecuencia la hemoglobinopatía SC (HSC) y la
talasemia.
Las manifestaciones clínicas más comunes de estas entidades son: anemia,
ictericia, crisis vasooclusivas (CVO) dolorosas recurrentes e infecciones bacterianas. En
el niño se puede producir el síndrome mano-pie y una causa importante de morbilidad y
también de mortalidad es la crisis de secuestro esplénico. El síndrome torácico agudo

36 (STA) es un motivo muy frecuente de ingreso y la complicación más grave es el
accidente vascular encefálico (AVE) de tipo oclusivo o hemorrágico. Otras
manifestaciones son: priapismo, úlceras maleolares, necrosis aséptica de la cabeza de
los huesos largos, retinopatía proliferativa y litiasis vesicular.
También existe oclusión microvascular subclínica que conduce en el paciente
adulto, a un daño orgánico crónico sobre todo al nivel pulmonar, cardíaco y renal.
Aunque se ha avanzado mucho en el conocimiento de la fisiopatología de la
drepanocitosis en las últimas décadas, todavía no se conoce completamente su historia
natural. Por otra parte, desde diferentes partes del mundo se comunican diferencias en
la expresión clínica de la enfermedad.
En la drepanocitosis se describen también lesiones hepáticas producidas por
falciformación crónica en los sinusoides, hepatitis viral, sobrecarga de hierro o una
combinación de estos factores. La crisis hepática que posiblemente se produce por una
vasooclusión intrahepática aguda, fue descrita por primera vez por Diggs en 1965.
Posteriormente se demostró que su curso clínico es muy variable desde la resolución
espontánea en poco días hasta la insuficiencia hepática y la muerte.
La AF se caracteriza por una anemia hemolítica grave, que aparece a los pocos
meses de nacer cuando la Hb S reemplaza a la Hb fetal, que predomina al nacer y
durante los primeros meses de vida. En los niños es frecuente encontrar una
esplenomegalia, que desaparece a medida que se producen infartos esplénicos
produciéndose una verdadera atrofia esplénica.
A la exploración física se aprecia un tinte ictérico conjuntival. La anemia es
hemolítica crónica. Los valores de Hb oscilan entre 6 y 8 gr/dl y se acompaña de una
intensa reticulocitosis. En el frotis de sangre se observan drepanocitos, que son claves
en el diagnóstico. Este se confirma con la electroforesis de Hb en medio alcalino y en
agar citrato a pH ácido. La hemoglobina fetal en los homocigotos se encuentra elevada
en proporción variable y parece actuar como mecanismo protector impidiendo la
falciformación.

37
En la anemia drepanocítica son frecuentes dos tipos de complicaciones:
– Crisis vasculares oclusivas o crisis de dolor: por acumulación de drepanocitos que
determina éstasis arterial e infartos. Las crisis vasculares se inician bruscamente,
con intenso dolor y fiebre. En los niños los lugares más frecuentes son los huesos de
las manos y los pies. Son frecuentes los procesos osteomielíticos por Salmonella. En
los adultos predominan los infartos pulmonares. Puede presentarse priapismo.
– Crisis aplásicas: por interrupción brusca de la producción de eritrocitos, secundario,
generalmente, a infecciones por parvovirus y deficiencias de ácido fólico.
El tratamiento específico no existe. Algunas precauciones y medidas generales
contribuyen a reducir el número de crisis, por ejemplo, evitar los cambios de
temperatura, la deshidratación y las infecciones a las cuales son muy susceptibles, por
la hipoesplenia que determinan los infartos repetidos del bazo.
Talasemias (Malcorra JJ. 2001; Fairbanks VF, 1980)
Bajo el nombre de “talasemia” se incluyen un grupo muy heterogéneo de
alteraciones congénitas cuya característica común es un defecto en la síntesis de una o
varias cadenas de globina normales. La disminución de la síntesis de cadenas alfa se
denomina alfatalasemia, la de cadenas beta, betatalasemia, la de cadenas delta y beta
simultáneamente, delta/betatalasemia, y así sucesivamente.
La herencia de la talasemia muestra un patrón autosómico dominante y su
frecuencia dentro del conjunto de la población mundial es muy elevada, presentando
una distribución que se correlaciona con las zonas donde existe o ha existido paludismo
endémico. Ello obedece al efecto protector que frente al parásito ejerce la
hemoglobinopatía, lo que significa una presión genética positiva de ésta sobre la
población afectada.
En algunos países de la cuenca mediterránea, pero especialmente en ciertas
zonas de Asia y África, la talasemia continua siendo un grave problema de salud
pública que obliga a implantar programas de prevención y diagnóstico prenatal. En
España, los estudios epidemiológicos más recientes han evidenciado una elevada

38 incidencia de los síndromes talasémicos con distribución geográfica irregular y marcada
heterogeneidad genotípica.
Fisiopatología: La disminución en la síntesis de un tipo de cadena globínica rompe
el equilibrio normal entre las cadenas alfa y beta y conduce a la acumulación
intracelular de una de ellas. Así, en la alfatalasemia se produce un exceso de cadenas
beta y en la betatalasemia un exceso de cadenas alfa. En ambos casos, se forman
precipitados intracelulares que son la causa de la destrucción precoz de los
eritroblastos antes de alcanzar la maduración completa (eritropoyesis ineficaz).
Así mismo, los eritrocitos que superan el trastorno madurativo, suelen presentar
también abundantes precipitados de cadenas globínicas en exceso que invariablemente
disminuyen su supervivencia en la circulación (hemólisis).
Aspectos moleculares: Desde el punto de vista molecular, las talasemias
obedecen a mutaciones de genes de la globina que, de alguna forma, alteran el
mecanismo de síntesis. En la alfatalasemia, tales mutaciones consisten en delecciones
de parte o todo un gen, y en otras, como en la betatalasemia, a sustituciones de un
nucleótido en los intrones (IVS), en la vecindad de éstos, en los exones, que alteran la
transcripción o la maduración del RNAm y dificultan o impiden la traducción.
Entre los métodos que más han contribuido al conocimiento del mecanismo
molecular de los síndromes talasémicos, destacan el análisis del DNA mediante
endonucleasas de restricción e hibridación con sondas marcadas radiactivamente (Sou-
thernblot) y la técnica de la reacción en cadena de la polimerasa (PCR).
ß- Talasemia
La betatalasemia obedece a una disminución en la síntesis de cadenas beta de
globina. La intensidad del déficit depende del grado de alteración genética y puede
variar desde una síntesis deficiente o parcial (ß+- tal) hasta una ausencia total de
síntesis (ß+- tal). La diferente expresividad clínica de la betatalasemia resulta de la
combinación de ambas posibilidades o de cada una de ellas con el gen normal.
Basándose en ello, la talasemia se clasifica clínicamente en 3 grandes grupos:

39 a) talasemia mayor o enfermedad de Cooley que corresponde a las formas de mayor
expresividad clínica (síndrome hemolítico crónico muy intenso con anemia grave y
esplenomegalia).
b) talasemia menor o rasgo talasémico que corresponde a formas de expresividad
clínica poco manifiesta o incluso ausente (talasemia mínima).
c) talasemia intermedia que corresponde a formas de expresividad clínica de diferente
intensidad, aunque siempre caracterizadas por un síndrome hemolítico moderado o
intenso con anemia y esplenomegalia.
Mecanismo molecular. El mecanismo molecular de la betatalasemia es heterogéneo,
aunque en prácticamente todos los casos obedece a cambios de una única base
nitrogenada (mutación puntiforme) que alteran la transcripción o el procesamiento
(maduración) del RNAm, disminuyendo (ß+-) o impidiendo (ß+-) su traducción.
Manifestaciones clínicas. Debido al elevado polimorfismo genético y a la existencia
de diversos mecanismos fisiopatológicos en el desarrollo de la anemia, la expresividad
clínica de la betatalasemia puede variar, desde una situación prácticamente
asintomática (talasemia minor), hasta la anemia intensa con fallecimiento del paciente
antes de alcanzarla edad adulta (talasemia mayor) con formas intermedias de
expresividad clínica muy variable (talasemia intermedia).
ß- Talasemia menor
Es la forma más frecuente de talasemia en nuestro medio y se caracteriza por una
seudopoliglobulia microcítica con anemia muy discreta o inexistente. Rara vez se
aprecia esplenomegalia. Debido a ello, el diagnóstico suele ser casi siempre casual y
facilitado por el empleo de autoanalizadores hematológicos que determinan
sistemáticamente el valor del VCM. La presencia de hipocromía hace que este trastorno
genético sea fácilmente tomado por una ferropenia con el consiguiente peligro de
someter al enfermo, si no se realiza el diagnóstico diferencial, a una prolongada, inútil y,
sobre todo, nociva sobrecarga de hierro.

40
El procedimiento más asequible para el diagnóstico de betatalasemia menor es la
práctica de una electroforesis de hemoglobinas, donde en sus formas más frecuentes
(heterocigotos ß+/ß y ß+/ß) se observa un aumento característico de la fracción HbA2
(3,8-7%) con HbF normal (< 2%). En prácticamente todos los casos, el diagnóstico de
betatalasemia menor se basa en la dosificación de la Hb A2 (aumento) y Hb F, y en un
estudio familiar.
�- Talasemia mayor
Se caracteriza por una expresividad clínica variable, pero generalmente intensa.
Su forma más grave es la anemia de Cooley. Esta se inicia a partir de los 6 meses del
nacimiento y se caracteriza por una intensa anemia, esplenomegalia, a veces gigante, y
hepatomegalia. La exploración física, muestra además de las visceromegalias,
alteraciones óseas, que se aprecian sobre todo en cráneo, que originen deformaciones
de su configuración, en especial en cara, configurando unos rasgos faciales
característicos.
El estudio radiológico muestra la imagen del llamado “cráneo en cepillo”. A veces
se observa intenso retraso del desarrollo, hecho que se puede evitar si se instaura
precozmente, en estos pacientes un régimen hipertrasfusional que procure mantener
los niveles de hemoglobina por encima de 10 gr/dl. El cuadro clínico se suele agravar
por las complicaciones debidas a la hemocromatosis (diabetes mellitus, miocardiopatía)
secundaria a la mayor absorción intestinal de hierro y efecto del régimen transfusional.
Estas complicaciones son precisamente las que constituyen la causa de muerte en
estos pacientes, casi siempre antes de los 25 años.
Diagnóstico
• El perfil hematológico nos muestra una anemia, por lo general, intensa, microcítica e
hipocroma.
• Examen morfológico de la sangre: intensa anisopoiquilocitosis, con hipocromía
acusada y abundante punteado basófilo. Es frecuente observar elementos inmaduros
de la serie roja.

41 • Los reticulocitos ligeramente aumentados, aunque nunca tanto como correspondería
al grado de anemia y eritroblastosis medular. Ello es un reflejo de la intensa
eritropoyesis ineficaz que invariablemente acompaña a esta enfermedad.
• El examen de médula ósea: hiperplasia eritroblástica de predominio ortocromático.
• La electroforesis de Hb evidencia un aumento de la Hb fetal que oscila entre el 60 y
el 98%
• Estudio familiar: comprobando la existencia de betatalasemia minor en los padres.
Tratamiento: Es esencialmente paliativo y consiste en la práctica de transfusiones
cuya periodicidad depende de la necesidad de mantener el nivel de hemoglobina. Las
transfusiones deben acompañarse de la administración de quelantes del hierro y
eventualmente de la práctica de una esplenectomía (ver mas adelante).
ß- Talasemia intermedia
En la betatalasemia intermedia el cuadro clínico es siempre manifiesto y se
caracteriza por una anemia de intensidad moderada, hemólisis crónica y
esplenomegalia, cuya gravedad no alcanza nunca la de la enfermedad de Cooley. En
general, estos pacientes no suelen requerir transfusiones ni es habitual observar en
ellos los rasgos propios de un déficit crónico de hemoglobina (retraso del desarrollo
pondoestatural y gonadal). Ocasionalmente, no obstante, pueden desarrollar
cardiomegalia, osteoporosis, fracturas espontáneas y artritis, y dado que existe
prácticamente siempre sobrecarga de hierro, es muy aconsejable la administración de
DFO.
α-Talasemia
La alfatalasemia, o disminución congénita de la síntesis de cadenas alfa, es otra
de las formas de talasemia frecuentes en nuestro medio, quizá aunque algo menos que
la betatalasemia. La alfatalasemia se caracteriza por la síntesis de un exceso de
cadenas gamma durante el período fetal y de cadenas beta después del nacimiento.
Durante el período fetal, las cadenas gamma en exceso forman homotetrámeros
(hemoglobina Bart) que después del nacimiento desaparecen y son sustituidos por

42 homotetrámeros ß �o hemoglobina H (HbH). Tanto la HbH como la hemoglobina Bart
(Hb Bart) pueden evidenciarse electroforéticamente. Al igual que la betatalasemia, la
alfatalasemia puede presentarse en diferentes formas clínicas que son expresión de un
diferente genotipo.
Mecanismo molecular. A diferencia de lo que sucede en la betatalasemia, en la
alfatalasemia predominan las delecciones de material genético sobre los cambios de
una sola base nitrogenada. En cualquier caso, la expresividad clínica dependerá de las
características de la mutación o del numero de genes afectados por la delección Las
formas moleculares más frecuentes de α+-talasemia son la deleción de 3,7 kb (α3,7-
talasemia) y la de 4,2 kb (α4,2-talasemia), aunque se han descrito también otras
delecciones cortas de 3,5 y 5,3 kb. La delección de 3,7 kb es especialmente frecuente
en el área mediterránea y en la población americana de raza negra, mientras que la
delección de 4,2 kb es más propia del sudeste asiático.
Manifestaciones clínicas: A diferencia de la ß-talasemia, la α-talasemia se manifiesta,
aún incluso, antes de nacer, ya que las cadenas α �forman parte de todas las
hemoglobinas tanto fetales como adultas. La expresividad clínica de la α-talasemia
depende de la naturaleza de la delección. En la práctica, las α -talasemias se clasifican
en 3 grandes grupos: α+-talasemia o tipo 2 (α-/αα,-α/- α); α0-talasemia o tipo 1 (—/αα, —
/—), y hemoglobinopatía H (α-/—).
1) α�+-talasemia o alfatalasemia tipo 2 obedece a la delección de un único gen alfa.
En su forma heterocigota constituye una forma de talasemia mínima, ya que por su
carácter asintomático pasa prácticamente siempre desapercibida. El estudio de la
síntesis de cadenas globínicas es normal, al igual que el patrón electroforético de
hemoglobinas, y el único criterio diagnóstico es una tendencia muy ligera a la
disminución del VCM. En realidad este tipo de portadores asintomáticos de α+-
talasemia solo se detectan al realizar el estudio familiar de pacientes con formas
homocigotas cuya expresividad clínica es superponible a la betatalasemia menor
(ligera anemia microcítica o seudopoliglobulia microcítica). En cualquiera de los
casos, la confirmación diagnóstica exige no sólo la realización de la síntesis in vitro

43
de cadenas de globina (alfa/beta <1), sino también un análisis del DNA mediante la
técnica de Southern para demostrar la delección del gen alfa.
2) α�0- talasemia o alfatalasemia tipo I obedece a la delección de 2 genes alfa y, en
su forma heterocigota, presenta una expresividad clínica superponible a la de
betatalasemia menor. A diferencia de ésta, no obstante, el patrón electroforético
muestra una HbA2 normal o incluso disminuida (1,5-2,5 %) y nunca se observa
aumento de HbF. El estudio de la síntesis de cadenas de globina muestra
prácticamente siempre una disminución del cociente alfa/beta (<1), y
ocasionalmente, puede aparecer precipitados intraeritrocitarios de HbH cuando los
eritrocitos se incuban en presencia de azul de cresilo brillante. Su forma homocigota
corresponde a la hidropesía fetal o α0-talasemia con Hb Barts. Esta forma de
alfatalasemia por delección de todos los genes de alfaglobina es incompatible con la
vida y se observa de forma casi exclusiva en países del sudeste asiático (China,
Cambodia, Tailandia y Filipinas), siendo rara en el Mediterráneo.
En general, constituye una causa de aborto hacia las 30 semanas del embarazo o
de muerte fetal poco después del nacimiento por anasarca fetoplacentaria (hidropesía
fetal).
Tratamiento de las hemoglobinopatías
Dos son las hemoglobinopatías que requieren un tratamiento regular: la talasemia
mayor y la drepanocitosis. El tratamiento es esencialmente paliativo y consiste en la
práctica de transfusiones periódicas, acompañadas de la administración de quelantes
del hierro y a veces de esplenectomía. Hoy en día el único tratamiento curativo es el
TMO alogénico, y está en fase experimental la manipulación genética.
Terapéutica transfusional 1.- Régimen clásico o de transfusión a demanda, y por el cual solo se transfunde
cuando la hemoglobina desciende por debajo de un determinado nivel (<70-80 g/l).

44 2.- Régimen de hipertransfusion, consistente en la práctica de tantas transfusiones
como sean necesarias para evitar que la hemoglobina descienda por debajo de 100 g/l.
Esta cifra es importante, ya que la reducción de eritropoyesis que comporta supone una
drástica disminución de la absorción de hierro intestinal. Una variante de la
hipertransfusión es la llamada “supertransfusion”, por la cual el hematocrito debe
mantenerse siempre por encima del 35% (Hb > 120 g/l). Un programa de
supertransfusión o hipertransfusión requiere, en general, la transfusión de 2-3
concentrados de hematíes cada 2-4 semanas.
Sus ventajas e inconvenientes con respecto al clásico son: Ventajas: mejor
desarrollo óseo, menor esplenomegalia, mejor calidad de vida; Inconvenientes: mayor
sobrecarga de hierro (hemocromatosis)
Esplenectomía: Las indicaciones de la esplenectomía en la talasemia vienen
dadas por: compresión sobre órganos vecinos, hiperesplenismo y aumento de los
requerimientos transfusionales.
Trasplante de medula ósea: Los resultados alcanzados hasta ahora señalan una
mortalidad del 10-20%, con una supervivencia media superior a 5 años del 75-90%. En
la indicación de TMO habrá que valorar las posibilidades de larga supervivencia con
tratamiento quelante y transfusional, y el riesgo importante inmediato de mortalidad que
comporta el TMO. Este es menos eficaz cuando existe valorable hepatomegalia y
fibrosis portal y ofrece precisamente sus mejores resultados en los casos que mejor
responden al tratamiento quelante y transfusional.
Manipulación genética: se halla aún en fase experimental. En la actualidad está
realizándose un gran esfuerzo en dos sentidos: activar la síntesis de cadenas gamma
(aumento de síntesis de HbF capaz de suplir el déficit de HbA) e inserción en las células
hematopoyéticas del gen beta normal. La activación de los genes gamma se ha logrado
con poco éxito práctico mediante administración del agente hipometilante 5-azacitidina
y esta intentándose de nuevo con hidroxiurea, (que al actuar directamente sobre la
BFU-E podría ejercer un efecto más prolongado). La inserción de genes constituye por
el momento uno de los objetivos más preciados, ya que las primeras experiencias

45 realizadas en animales demuestran que los genes de globina vehiculizados por un
retrovirus pueden ser transferidos (transfección) a células hematopoyéticas humanas.
Sobrecarga de Hierro (Altésa A y col, 2007).
Como contrapartida, el intenso régimen transfusional supone siempre un mayor
aporte de hierro al organismo y, por tanto, el peligro de una hemocromatosis que, por la
gravedad de sus complicaciones, puede ocasionar el fallecimiento del paciente casi
siempre antes de los 25 años de edad. Por ello, cualquier régimen transfusional se
acompaña de la administración de quelantes del hierro. Este gran aporte de hierro que
ocasiona daño orgánico, aunque se le reconoce que es un elemento imprescindible
para la salud, su exceso puede entrañar riesgos importantes.
Son pocos los estudios realizados a nivel internacional que analicen la distribución
y el metabolismo de hierro en la población. En España por ejemplo, la mayoría se han
centrado en población pediátrica o en mujeres en edad fértil, con el propósito de evaluar
la prevalencia de ferropenia y anemia ferropénica en poblaciones de riesgo.
Tradicionalmente se ha prestado poca atención a la población adulta en general y a los
ancianos en particular, en quienes la prevalencia de sobrecarga férrica puede ser
mayor.
El descubrimiento de las mutaciones del gen HFE ligadas a la hemocromatosis
hereditaria y del síndrome de sobrecarga férrica y dismetabolismo, ha elevado el interés
de los investigadores por el tema de la sobrecarga férrica.
Los datos sorprendentes del estudio publicado por Altésa y colaboradores (2007)
son relativos a la sobrecarga de hierro en la población, donde el 9,3% de los adultos
(11,7% en varones jóvenes y 20,4% en mayores de 50 años), presentaron sobrecarga
férrica, mas aún cuando un 1,6% de la población general (2,5% de varones jóvenes y
3,9% de mayores de 50 años) presentaban valores de ferritina superiores a 500 μg/l.
Este fenómeno afectó también a las mujeres. El 1,2% de las mujeres jóvenes y el 9,4%
de las mayores de 50 años, presentaron sobrecarga férrica. Otros estudios en países
occidentales han observado el mismo fenómeno.

46
En la evaluación del estado férrico de una población de 1.016 ancianos, con
edades comprendidas entre los 67 y 96 años y pertenecientes al Framingham Heart
Study, un 3% presentó depleción férrica y el 13%, sobrecarga (Fleming y col, 2001;
Milman y col, 2002; Milman y col, 2003) demuestran que la sobrecarga férrica en los
varones daneses se incrementó entre 1984 y 1994 del 11,3 al 18,9%, y en las mujeres
danesas posmenopáusicas, del 2,4 al 5,5%.
Una posible explicación de la elevada frecuencia de sobrecarga férrica sería la
presencia de hemocromatosis hereditaria. No obstante, esta enfermedad, según
estudios realizados en España el mismo lugar del trabajo Altesa (2007), afecta
solamente a 1 de cada 1.000 personas, y por tanto sólo podría explicar un pequeño
porcentaje de los casos. Una segunda explicación de los elevados índices de ferritina,
podría ser la alta prevalencia de infección por virus de la hepatitis C en la población. En
tercer lugar, en algunas series de pacientes con sobrecarga férrica el llamado síndrome
de sobrecarga férrica ligada a dismetabolismo ha demostrado ser un factor prevalente y
finalmente, las anemias hemolíticas como la AF y las Talasemias, entre otras, por el
régimen transfusional (Altes y col, 2003).
Sea cual sea la causa del incremento en la sobrecarga férrica, debe recordarse los
numerosos trastornos orgánicos en los que ésta parece estar implicada y que pueden
afectar gravemente a la salud de las personas, por lo que constituye un importante dato
para los responsables de la salud pública Hasta el momento, el método de cribado de
elección para la hemocromatosis, sobre todo la hereditaria, consiste en determinar el
índice de saturación de la transferrina y estudiar exhaustivamente a las personas con
índice superior al 45% en 2 ocasiones.
Mientras que en sectores específicos de la población (con alta prevalencia de
anemia ferropénica) es necesario mantener la vigilancia en cuanto al déficit de este
metal, en el resto de la población la enfermedad más frecuente es la relacionada con la
sobrecarga de hierro. Deben implementarse por tanto programas específicos para
detectar esta anomalía, analizar sus causas y los motivos de su progresión, a fin de
adoptar medidas correctoras. Respecto a este último punto, es injustificable el

47 «enriquecimiento» con hierro mineral de los alimentos de uso general, no destinados
específicamente a población con riesgo de ferropenia, con objetivos comerciales.
Metabolismo del hierro
El hierro es el elemento traza esencial más abundante del organismo y es
necesario para la proliferación y diferenciación celular, el transporte y la fijación de
oxígeno, la síntesis de ADN y de proteínas mitocondriales necesarias para el transporte
de electrones y la producción de energía la regulación de la expresión
postranscripcional de diferentes genes
El hierro posee la capacidad de aceptar o donar un electrón fácilmente (potencial
redox) y puede existir en las dos formas interconvertibles Fe (II) y Fe (III). La forma
oxidada de hierro Fe (III) es muy poco soluble a pH fisiológico (10-18 mol/L) y por otra
parte, el exceso de la forma reducida Fe (II) es potencialmente tóxico debido a la
formación de radicales libres de oxígeno: Fe (II) + H2O2 ® Fe (III) + OH- + OH. (Reacción de Fenton). Las especies reactivas de oxígeno son citotóxicas a nivel de las
membranas celulares (lipoperoxidación), proteínas y ácidos nucleicos (Lieu PT, 2001).
Debido al potencial tóxico del exceso de hierro intracelular, el balance del hierro –
captación, transporte, almacenamiento y utilización– está altamente regulado a nivel
celular (existencia de numerosas proteínas reguladoras) y del organismo (control de la
absorción intestinal, el almacenamiento hepático y la liberación de hierro por los
macrófagos) (Lieu PT, 2001, Hentze MW, 2004, Roy CN, 2001, Sherwood RA, 1998,
Fleming RE, 2005)
Como consecuencia de su complejidad, la prevalencia de los desórdenes de la
homeostasis del hierro es muy elevada, incluye diferentes manifestaciones clínicas y
abarca todo el espectro desde la deficiencia de hierro y la anemia ferropénica hasta la
sobrecarga férrica (hemocromatosis) (Lieu PT, 2001, Sherwood RA, 1998, Worwood M,
1997).

48
El contenido total del hierro del organismo, 3 a 4 g, se distribuye en tres
compartimentos: funcional, de depósito y plasmático (de transporte). Aproximadamente
el 60-70% del contenido de hierro del organismo está constituido por hierro funcional
que se localiza esencialmente en la hemoglobina de los hematíes maduros (circulación)
y precursores eritroides (médula ósea), y el 10-15% en la mioglobina (músculo
esquelético) y en los citocromos y enzimas de diferentes tejidos. Entre el 20-30% del
hierro total es almacenado en las células del parénquima hepático y en los macrófagos
del sistema reticuloendotelial en forma de ferritina y hemosiderina (hierro de depósito).
Aproximadamente 3 mg de hierro (<0.1% del hierro del organismo) circulan en el
plasma como un pool de hierro intercambiable unido a la transferrina. La transferrina
facilita la distribución y el intercambio celular del hierro en los tejidos mediante la
interacción con los receptores de transferrina celulares (Lieu PT, 2001; Hentze MW,
2004; Worwood M, 1997). Además la transferrina aumenta la solubilidad del Fe (III) en
condiciones fisiológicas, y previene la formación de radicales libres de oxígeno
citotóxicos.
Diariamente se absorben 1-2 mg de hierro de la dieta por los enterocitos
duodenales para compensar las pérdidas habituales. Debido a la limitada disponibilidad
de hierro a partir de la dieta, el organismo posee un sistema de reciclaje muy eficiente
para la conservación del hierro. Los macrófagos del sistema reticuloendotelial
proporcionan la mayor parte del hierro reutilizado a partir de la degradación de la
hemoglobina de los hematíes viejos (eritrofagocitosis; vida media 120 días) y la cesión
del Fe (III) a la transferrina.
En cuanto a la deficiencia de hierro es la alteración nutricional más común y
ampliamente distribuida, es la principal causa de anemia, especialmente en niños en
edad preescolar y adolescentes, mujeres en edad fértil y ancianos. La deficiencia de
hierro se relaciona con aporte dietario de hierro inadecuado, malabsorción intestinal del
hierro, requerimiento fisiológico aumentado de hierro (períodos de crecimiento rápido),
pérdidas excesivas o crónicas de sangre, e infestaciones parasitarias.

49
La deficiencia de hierro comprende tres etapas secuenciales (Pérez Surribas y
Viedma JA, 2006):
• Depleción de los depósitos de hierro, caracterizada por la presencia de
concentraciones plasmáticas de ferritina bajas.
• Deficiencia de hierro funcional. La eritropoyesis disminuye debido al aporte
inadecuado de hierro a los precursores eritrocitarios de la médula ósea (eritropoyesis
ferropénica por deficiencia relativa de hierro). Característicamente se observa
ferrropenia, índice de saturación de transferrina bajo, aumento de la concentración de
transferrina y de receptor soluble de transferrina.
• Anemia ferropénica, deficiencia absoluta de hierro de depósito. La concentración
sanguínea de hemoglobina se halla significativamente disminuida. La anemia
ferropénica se caracteriza por hipocromía, microcitosis, y anisocitosis. La
concentración plasmática de ferritina es baja, si no coexiste inflamación.
La anemia de las enfermedades crónicas, la segunda forma de anemia más
prevalente, se asocia a inflamación crónica, y se caracteriza por una distribución
inadecuada del hierro en el organismo (acúmulo de hierro en macrófagos y
eritropoyesis ferropénica a pesar de existir depósitos de hierro). En la mayoría de los
casos se debe a infección, inflamación (incluyendo conectivopatías) y neoplasia. El
mecanismo es complejo y depende de la activación crónica de células inflamatorias y la
producción excesiva de citocinas, principalmente proinflamatorias (IL-1b, IL-6, TNF-a e
IFN-g) (6).
Las citocinas proinflamatorias estimulan la captación y almacenamiento de hierro
en los macrófagos del sistema reticuloendotelial (acumulación de ferritina y
hemosiderina), inhiben la proliferación y diferenciación de células progenitoras eritroides
(efectos inhibidores de IFN-g, TNF-a e IL-1, inducción de apoptosis por TNF-a), y la
respuesta de la médula ósea a la eritropoyetina, y reducen la vida media de los
hematíes. La expresión aumentada en hepatocitos de hepcidina, proteína de fase
aguda cuya síntesis es estimulada por IL-6 y endotoxina, produce en modelos
experimentales anemia, inhibiendo la absorción duodenal de hierro. Las citocinas
antiinflamatorias (IL-4, IL-10 e IL-13) favorecen la retención de hierro en los macrófagos

50 activados y participan en la inducción de ferropenia e hiperferritinemia de las
enfermedades crónicas inflamatorias (Weiss G, 2005; Tilg H, 2002)
La anemia de la insuficiencia renal crónica representa un caso especial de anemia
asociada a enfermedades crónicas. La anemia, normocítica e hipocroma, resulta
principalmente del déficit de eritropoyetina (hu-EPO) debido a la reducción de la síntesis
por la enfermedad renal y a los efectos inhibitorios de las toxinas urémicas sobre la
proliferación de células progenitoras eritroides (Weiss G, 2005, Goodnough LT, 2000).
La hemocromatosis hereditaria incluye un grupo heterogéneo de desórdenes
autosómicos recesivos caracterizados clínicamente por la presencia de sobrecarga
férrica y depósito de hierro con afectación de diferentes órganos, principalmente
hígado, glándulas endocrinas y corazón (Pietrangelo A, 2004).
Se han definido cuatro tipos diferentes de hemocromatosis hereditaria debido a
mutaciones de genes que codifican proteínas reguladoras de la homeostasis del hierro.
• Tipo 1 Hemocromatosis hereditaria clásica (HFE, 6p21.3)
• Tipo 2 Hemocromatosis hereditaria juvenil
Subtipo A (hemojuvelina, 1q21)
Subtipo B (hepcidina, 19q13.1)
• Tipo 3 Hemocromatosis hereditaria relacionada con TfR2 (7q22)
• Tipo 4 Sobrecarga de hierro relacionada con ferroportina (2q32)
La forma clásica de hemocromatosis hereditaria es la más común. Se debe a una
mutación del gen HFE, localizado en el cromosoma 6p21.3. En la mayoría de los casos
la mutación consiste en la simple sustitución de un residuo de tirosina por cisteína en la
posición 282 de la proteína HFE (C282Y). Se han descrito otras mutaciones menos
frecuentes, como la mutación en la que ácido aspártico reemplaza a histidina en la
posición 63 (H63D).
La hemocromatosis adquirida o secundaria se observa en las talasemias y
anemias hemolíticas, la anemia sideroblástica, y la siderosis transfusional.

51 Magnitudes proteicas en la exploración del metabolismo del hierro (Pérez Surribas
y Viedma JA, 2006) Transferrina.
La transferrina es una glucoproteína (contenido variable de cadenas laterales de
carbohidratos, hasta 6% de la masa molecular) de síntesis principalmente hepática, con
movilidad electroforética en la zona ß1-globulina. El gen de la transferrina se localiza en
el cromosoma 3q21. La molécula de transferrina consiste en una cadena polipeptídica
de 679 aminoácidos (79,6kDa) con dos lóbulos homólogos N-terminal y C-terminal,
conteniendo cada uno un sitio de fijación de elevada afinidad para hierro Fe (III). Se han
descrito más de 20 variantes genéticas de transferrina.
La transferrina existe en la circulación como apotransferrina, y formas mono-
diférricas. El receptor de transferrina tiene mayor afinidad por la forma diférrica. La
función biológica de la transferrina es fijar y transportar hierro Fe (III) extracelular. La
transferrina recibe los átomos de Fe (III) de enterocitos duodenales, células del
parénquima (hepatocitos), o macrófagos del sistema reticuloendotelial, y los libera en la
médula ósea y en diferentes tejidos para su utilización, o en el hígado para su
almacenamiento, a través de la interacción con receptores específicos de transferrina
((Sherwood RA, 1998; Fleming RE, 2005; Worwood M. 1997).
La concentración de transferrina en suero o plasma está disminuida en:
• Respuesta de fase aguda (inflamación, infección, necrosis, trauma, cirugía)
• Neoplasia
• Enfermedad hepática
• Enfermedad renal (síndrome nefrótico, diálisis, insuficiencia renal crónica)
• Malnutrición (marcador de malnutrición en ausencia de inflamación)
• Sobrecarga de hierro (hemocromatosis hereditaria)
La concentración de transferrina en suero o plasma está aumentada en:
• Deficiencia de hierro
• Embarazo y terapia con estrógenos

52
La principal indicación de la determinación de la concentración plasmática de
transferrina es el diagnóstico diferencial de la anemia (en combinación con ferritina y el
índice de saturación de la transferrina): la concentración de transferrina aumenta en la
anemia ferropénica y está baja en la anemia de las enfermedades crónicas sin
componente ferropénico (dentro de los valores de referencia si hay ferropenia asociada)
Los principios de medida más utilizados para la determinación de la transferrina
son la inmunonefelometría y la inmunoturbidimetría. Existe un material de referencia
internacional certificado (RPPHS-CRM 470) con valores asignados para 14 proteínas
incluyendo la transferrina.
La capacidad total de fijación de hierro por el suero es la masa de hierro máxima
que puede transportar un volumen determinado de suero. La capacidad total de fijación
de hierro por el suero es una medida indirecta de la concentración de transferrina. El
método clásico de Ramsay se basa en la determinación de la concentración de hierro
que es capaz de fijar el suero tras la saturación con hierro Fe(III) y la eliminación del
exceso mediante adsorción con carbonato de calcio o magnesio (Ramsay WNM, 1957).
La capacidad de fijación de hierro por la transferrina es la masa de hierro teórica
que transporta la transferrina presente en un volumen determinado de suero. Se trata
de una estimación a partir de la concentración plasmática de transferrina. En el cálculo
de la capacidad de fijación del hierro por la transferrina (CFTf) se considera la
proporción de dos iones férricos por una molécula de transferrina: CFTf (μmol hierro/L)
= Transferrina (μmol/L) x 2. Dado que el peso molecular de la transferrina es de 79,6
kDa, la fórmula queda como: CFTf (μmol hierro/L) = Transferrina (g/L) x 25,1
El coeficiente de saturación es el cociente expresado como porcentaje entre la
sideremia y la capacidad total de fijación de hierro por el suero.
El índice de saturación de transferrina (ISTf) es el cociente expresado como
porcentaje entre la sideremia y la capacidad de fijación del hierro por la transferrina.
ISTf = Hierro sérico total/CFTf x 100.

53 Ferritina
Las ferritinas constituyen una amplia superfamilia altamente conservada de
proteínas de almacenamiento de hierro. La estructura molecular común es la de una
esfera hueca formada por una cubierta proteica globular formada por 24 subunidades
unidas por enlaces no covalentes (apoferritina) de aproximadamente 450 kDa y 120 A
de diámetro, y una cavidad central de 80 A de diámetro capaz de contener hasta 4500
átomos de hierro (III) en forma de cristales de [Fe(O)OH].
Las moléculas de apoferritina contienen dos tipos de subunidades estructurales
denominadas H (Heavy, heart) de 21 kDa y punto isoeléctrico ácido (4.8 - 5.2), y L
(Light, liver) de 19.7 kDa y punto isoeléctrico más básico (5.3 - 5.8), codificadas
respectivamente en los cromosomas 11 y 19. Ambas subunidades presentan
aproximadamente 55% de homología en la secuencia de aminoácidos. Existen
diferentes heteropolímeros cuyo contenido de cadenas polipeptídicas H y L varía de
acuerdo con el órgano y los requerimientos de hierro. Las cadenas H confieren
actividad de ferroxidasa al heteropolímero y las cadenas L proporcionan sitios de
nucleación para la fijación de hierro.
Las propiedades electroforéticas, inmunológicas y metabólicas de las isoferritinas
aisladas de diferentes tejidos dependen esencialmente del cociente H/L de la molécula.
Las ferritinas ricas en subunidades H poseen elevada actividad de ferroxidasa y
almacenan hierro de forma limitada (<1000 átomos de hierro III por molécula de
ferritina). Las ferritinas ricas en subunidades L, características de tejidos que almacenan
hierro como el hígado y el bazo, contienen >1500 átomos de hierro/molécula de
ferritina.
La mayor parte de las células del organismo contienen ferritina en el citosol,
siendo especialmente abundante su expresión en las células relacionadas con la
síntesis de hemoglobina (eritroblastos y reticulocitos), con su degradación
(macrófagos), o con su reserva (hepatocitos) (6,12). La ferritina citosólica es producida
por el retículo endoplásmico liso y no está glucosilada. La síntesis de ferritina

54 (subunidades H y L) es regulada a nivel posttranscripcional en el citosol y depende de
la concentración de hierro libre intracelular.
La ferritina plasmática es sintetizada por el retículo endoplásmico rugoso y es
glicosilada en el aparato de Golgi antes de su liberación. La ferritina plasmática es un
homopolímero L que contiene exclusivamente subunidades de tipo L (60-80%
glicosiladas) y posee un contenido de hierro muy bajo. La mayoría de las células
producen ferritina y secretan una proporción de ferritina glicosilada al plasma, por lo que
la concentración de ferritina plasmática refleja la cantidad de ferritina del organismo y
por tanto los depósitos de hierro (Carrondo MA, 1957; Jensen P-D, 2004).
Un tercer tipo de ferritina, ferritina mitocondrial, ha sido recientemente
caracterizado (Jensen P-D, 2004). La ferritina mitocondrial está formada por
subunidades de 22 kDa de tipo H codificadas por un gen en el cromosoma 5q23.1. La
ferritina mitocondrial posee actividad ferroxidasa (citoprotector en anemias
sideroblásticas), y además tiene una función moduladora en el tráfico de hierro desde el
citoplasma a las mitocondrias, y en la síntesis del grupo hemo (Levi S, 2001; Napier I,
2005).
En ausencia de inflamación, la concentración plasmática de ferritina correlaciona
estrechamente con los depósitos de hierro del organismo (Sherwood RA, 1998;
Worwood M, 1997)
La concentración de ferritina plasmática está disminuida en:
• Deficiencia de hierro
• Embarazo
• Pérdidas crónicas de sangre
La concentración de ferritina plasmática está aumentada en:
• Enfermedad hepática
• Respuesta de fase aguda (inflamación, infección)
• Neoplasias (linfoma, leucemia, carcinomas)
• Anemia de enfermedad crónica

55 • Insuficiencia renal crónica
• Talasemia
• Anemia sideroblástica
• Hemocromatosis hereditaria
• Hiperferritinemia hereditaria con cataratas congénitas
Las indicaciones para la medida de la concentración plasmática de la ferritina son
la detección de deficiencia de hierro y la monitorización del tratamiento. La ferritina es
una proteína de fase aguda que aumenta significativamente en procesos inflamatorios,
infección, hepatopatías (incluyendo hemocromatosis hereditaria), y ciertas neoplasias.
El aumento de la concentración de ferritina en enfermedades crónicas, independiente
de los depósitos de hierro constituye la principal limitación del uso de la ferritina para la
detección de deficiencia de hierro. Aunque la ferritina no es un buen marcador para la
detección de sobrecarga férrica, resulta útil para monitorizar el tratamiento con sangrías
de la hemocromatosis hereditaria.
En la determinación de la concentración plasmática de ferritina se utilizan
inmunoanálisis con diferentes principios de medida. Existe un material de referencia de
la Oficina Comunitaria de Referencia (BCR) preparado originalmente a partir de ferritina
humana del hígado o el bazo, certificado por la Organización Mundial de la Salud, y
distribuido por el Instituto Nacional de Patrones Biológicos y Control (NIBSC). El
material de referencia empleado actualmente se obtuvo a partir de ferritina
recombinante: NIBSC 94/572.
Receptor soluble de transferrina (Pérez Surribas y Viedma JA, 2006)
El receptor de transferrina TfR1 es una glucoproteína transmembrana de
estructura homodimérica, de masa molecular 190 kDa. Cada monómero contiene 760
aminoácidos (95 kDa). El extremo aminoterminal está en el dominio citoplasmático. El
dominio extracelular contiene dos puentes disulfuro (posición 89 y 98) que unen los dos
monómeros. Cada molécula de receptor puede fijar dos moléculas de transferrina.
Como la transferrina, el gen que codifica el receptor de transferrina TfR1 está localizado
en el cromosoma 3q26.

56
El receptor de transferrina TfR1 se localiza en la mayoría de las células, con la
excepción de los hematíes maduros. La mayor expresión de receptor de transferrina
TfR1 ocurre en los tejidos que sintetizan hemoglobina (precursores eritroides en la
médula ósea), y también en células normales en fase de división rápida, en la placenta,
y en tejidos neoplásicos.
El receptor de transferrina TfR1 es una molécula esencial para la captación celular
de hierro. La constante de asociación de la transferrina diférrica es muy superior a la de
las formas monoférricas y apotransferrina (Fleming RE, 2005).
Mediante un mecanismo de endocitosis, los receptores de transferrina unidos al
ligando (transferrina diférrica) son incluidos en un endosoma especializado, donde
mediante la acción de una bomba de protones (ATP-asa dependiente) se produce un
descenso en el pH que produce cambios conformacionales en las proteínas que
resultan en la liberación del hierro de la transferrina en el citosol. En las células
eritroides medulares la mayor parte del hierro liberado se utiliza para la síntesis de
hemoglobina, mientras que el exceso se deposita en la ferritina. La apotransferrina
resultante y el receptor son reciclados y utilizados en nuevos ciclos de fijación y
captación de hierro.
El receptor de transferrina TfR2, (7q22), presenta moderada homología con TfR1.
La homología de aminoácidos del dominio extracelular es del 45%. La expresión de
TfR2, a diferencia de TfR1, es específica del tejido hepático y duodenal, y no hay
mecanismo de regulación postranscripcional. Estudios muy recientes indican que el
hígado es el principal regulador de la absorción de hierro de la dieta y de la liberación
del hierro almacenado. Las moléculas de TfR2 actúan como sensores de la
concentración plasmática de transferrina diférrica. La expresión de TfR2 depende
directamente de la concentración de transferrina diférrica (holotransferrina),
independientemente de la presencia de anemia o de sobrecarga férrica hepática. Existe
evidencia de que en el hígado TfR2, hemojuvelina y HFE, son reguladores de la síntesis
y secreción de hepcidina, hormona que regula la absorción intestinal de hierro, y
actuando sobre hepatocitos y macrófagos, el almacenamiento y liberación de hierro.

57
El receptor soluble de transferrina es una forma truncada del receptor de
membrana que resulta de la pérdida de los dominios citoplasmático y transmembrana, y
que se origina mediante proteolisis entre los aminoácidos 100 y 101. Se trata de una
forma monomérica de masa molecular 85 kDa que forma un complejo con una molécula
de transferrina.
Los precursores medulares eritroides (eritroblastos) constituyen la principal fuente
de receptor soluble de transferrina (70-80% del total). Existe una buena correlación
entre la concentración sérica de receptor de transferrina y la actividad proliferativa
eritropoyética medular.
La concentración de receptor soluble de transferrina aumenta rápidamente en la
eritropoyesis ferropénica, y no está afectada por inflamación, infección, hepatopatías,
terapia con estrógenos o embarazo. La determinación de la concentración de receptor
soluble de transferrina en suero es útil para la detección de la depleción de hierro
funcional y para el diagnóstico diferencial entre la anemia ferropénica y la anemia de
enfermedades crónicas, y para evaluar la presencia de ferropenia en enfermedades
inflamatorias, infecciones, y neoplasias.
La concentración de receptor soluble de transferrina está disminuida en:
• Anemia hipoplástica
• Aplasia medular (post-quimioterapia)
• Hemocromatosis hereditaria
La concentración de receptor soluble de transferrina está aumentada en:
• Deficiencia de hierro funcional y anemia ferropénica
• Anemia hemolítica
• Eritropoyesis hiperproliferativa (talasemia, anemia megaloblástica)
La determinación de la concentración del receptor soluble de transferrina utiliza
inmunoanálisis con diferente principio de medida. Actualmente no existen materiales de
referencia certificados para el receptor soluble de transferrina, ni tampoco consenso
entre los fabricantes sobre la obtención de calibradores. La disparidad de valores de
referencia y valores discriminantes para la concentración de receptor soluble de

58 transferrina, dependiendo del calibrador y el método empleado, dificulta su aplicación
clínica en la práctica, y obliga a que el seguimiento para un mismo paciente se realice
en un único laboratorio.
Índices que relacionan receptor soluble de transferrina y ferritina.
Se utilizan dos índices: el índice receptor soluble de transferrina/log ferritina y el
índice receptor soluble de transferrina x 100/ferritina. Estos índices son especialmente
útiles para diferenciar la anemia ferropénica de la anemia de enfermedades crónicas y
para detectar deficiencia de hierro en pacientes con anemia de enfermedades crónicas.
Las anemias crónicas hereditarias, las transfusiones y la sobrecarga de hierro (Cano-Castellanos R y c63, 2009)
Entre las anemias crónicas hereditarias las mas frecuentes son la anemia
drepanocítica y los síndromes talasémicos, entre otros, cursan con requerimiento
transfusional elevados y consecuentemente, con el peligro potencial de desarrollar
sobrecarga de hierro.
La sobrecarga de hierro es el resultado de la incapacidad del organismo para
eliminar el hierro acumulado, podría evitarse en buena medida. Sin embargo, en
múltiples ocasiones, la atención que demanda el padecimiento principal impide que el
clínico estime suficientemente el riesgo de sobrecarga de hierro.
El hierro de la sangre transfundida es procesado inicialmente por los macrófagos,
que digieren los eritrocitos senescentes y retornan el hierro a la transferrina del plasma.
Esta carga de hierro transfusional puede saturar la transferrina y llevar a la formación de
“hierro no unido a transferrina” en el plasma; éste es tomado por las células del
parénquima hepático y depositado como ferritina y hemosiderina histológicamente
visibles. La mayoría del hierro depositado en esta forma se acumula en el hígado.
El hierro puede ser reducido de férrico (Fe+3) a ferroso (Fe2+), lo que le permite
catalizar la formación de radicales hidroxilo (altamente reactivos), que pueden causar
daño oxidativo y afectar lípidos, proteínas y moléculas de ADN. Probablemente, el
efecto sobre los lípidos juegue el principal papel del daño oxidativo mediado por hierro.
El resultado final es la descomposición de moléculas de lípidos, con el con comitante

59 efecto en la integridad de los organelos que pueden llevar a la muerte celular. Otro
efecto de los radicales es la producción aumentada del factor transformante de
crecimiento β-1 (TGF-β1), que conduce a una síntesis aumentada de colágena y a
fibrosis. En resumen, el daño oxidativo inducido por hierro puede producir muerte
celular o fibrosis.
En cuanto a la transfusión de concentrados eritrocitarios, si se administra un
volumen ≥120 mL/kg puede ocasionar sobrecarga de hierro que se correlaciona con
niveles de ferritina en suero iguales o mayores de 1 000 μg/L. Todos los pacientes con
sobrecarga de hierro deben ser vigilados en centros especializados, donde se pueda
evaluar la toxicidad orgánica, además de brindar educación para la salud a los
pacientes y apoyo general; pero en países como el nuestro, una parte significativa de la
población no siempre tiene acceso a este tipo de seguimiento.
Para establecer el diagnóstico de sobrecarga de hierro es necesario conocer la
concentración del metal en diferentes órganos y evaluar el funcionamiento del corazón,
hígado y glándulas endocrinas. La medición bioquímica cuantitativa del hierro no
hemínico en biopsias hepáticas constituye el método más exacto para evaluar la
magnitud de la sobrecarga de hierro, así como para guiar el tratamiento y, aunque no
se le puede considerar una prueba simple, tampoco es muy difícil de realizar por
médicos experimentados, ya que se trata de un procedimiento seguro. La muestra debe
ser de tamaño adecuado (1 mg de peso seco) y libre de cirrosis o lesiones focales del
hígado. La medición se realiza por espectrometría de absorción atómica en laboratorios
de referencia.
En algunos programas se recomienda realizar biopsias hepáticas al inicio del
tratamiento quelante y posteriormente cada dos años. La mejor indicación para iniciar
terapia quelante es la obtención de una concentración de hierro no hemínico mayor de
7 mg/g de peso seco de hígado.
Dentro de las mediciones indirectas, usualmente el empleo de dos o más
parámetros proporciona una buena aproximación de la cantidad total de hierro
acumulada. El hierro en suero se encuentra siempre elevado y la saturación de

60 transferrina (completamente saturada) correlaciona razonablemente bien con la ferritina.
Ésta es la principal proteína de almacenamiento intracelular de hierro y circula en el
plasma en pequeñas cantidades.
Se desconoce la función fisiológica de la ferritina secretada al plasma, pero parece
tener una relación logarítmica con las reservas de hierro del organismo. La ferritina en
plasma, considerada reactante de fase aguda, y la deficiencia de ácido ascórbico,
infección o inflamación crónica o aguda y concentración elevada de eritropoyetina
pueden alterar la relación entre ferritina sérica y las reservas de hierro en el organismo.
También se puede elevar por la liberación de ferritinas tisulares, ocasionada por daño al
hígado o a otros tejidos ricos en ferritina, así como por disfunción hepática.
Los pacientes con anemia drepanocítica que reciben transfusiones frecuentes
tienen un mayor riesgo de presentar varias condiciones que afectan la relación de
ferritina y reservas de hierro, especialmente infecciones crónicas y agudas, respuesta
inflamatoria a los infartos de la microvasculatura, enfermedad hepática y anemia
hemolítica crónica con hiperplasia eritroide. No obstante lo anterior, las mediciones
seriadas de ferritina sérica siguen siendo un método confiable y sencillo para evaluar la
tendencia de la sobrecarga de hierro y la eficacia de la terapia de quelación.
El receptor soluble de la transferrina (RTf) en plasma deriva de la masa total de
transferrina celular con 80% o más proveniente de la médula eritroide. Aunque algunos
informes indican que la concentración del RTf en plasma está disminuida en presencia
de sobrecarga de hierro, no se ha descrito ninguna relación cuantitativa entre la
magnitud de la sobrecarga de hierro y la concentración del RTf.
Un método no invasivo para la evaluación de la sobrecarga de hierro es el
Dispositivo de Interferencia Cuántica de Superconducción (SQUID por sus siglas en
inglés: Superconducting Quantum Interference Device). Sin embargo, este dispositivo
no es un recurso disponible en nuestro país; por ahora sólo cuentan con él cinco
centros de todo el mundo. Además, hay que considerar que en algunos estudios no se
encontró correlación entre los resultados obtenidos con el SQUID, los niveles de
ferritina sérica y la concentración de hierro en hígado.

61
La técnica o secuencia de pulso que ofrece mayor representatividad en el estudio
del sistema músculo esquelético es la imagen potenciada en T2*; así, la resonancia
magnética (RM T2*) puede usarse para valorar la carga de hierro en el hígado, corazón
y otros órganos como el páncreas y, en consecuencia, sustituir procedimientos
invasivos como la biopsia cardiaca y hepática. Las evaluaciones funcionales del
miocardio pueden realizarse periódicamente, ya que la sobrecarga de hierro al
miocardio es la causa más común de muerte por esta condición.
Una vez identificado un paciente con altos requerimientos transfusional es
necesario tratar de prevenir la sobrecarga de hierro, lo que se logra con el
reconocimiento de la enfermedad de base y la institución del tratamiento específico.
Cuando la sobrecarga de hierro ya está instalada es necesario tomar medidas dirigidas
a eliminar el hierro excedente.
Deferoxamina. Es un ácido trihidroxámico producido naturalmente por
Streptomyces pilosus que tiene una gran afinidad por el hierro, al que se une en una
relación de 1:1, para formar un complejo hexadentado de hierro. La deferoxamina es
pobremente absorbida por el tracto digestivo y tiene una vida media muy corta (20 min),
por lo que debe administrarse por vía parenteral. Su eficacia para reducir el daño
orgánico inducido por la sobrecarga de hierro se demostró desde la década de los años
setenta, utilizando como modelo a los pacientes talasémicos. Los resultados mostraron
que su administración por un período de 52 a 83 meses reducía significativamente el
riesgo hepático de fibrosis por acumulación de hierro, y también disminuía la incidencia
de insuficiencia cardiaca al revertir los cuadros asociados como la arritmia.
Para la vía de administración subcutánea se recomiendan 20-40 mg/kg/día en
infusión continua de 10-12 horas, de preferencia nocturna, con bomba de infusión,
durante cinco días a la semana, hasta lograr niveles de ferritina sérica ≤500 μg/L.
Igualmente, para la vía intravenosa se utiliza 20-40 mg/kg/día, disueltos en 1 000
mL de solución glucosada al 5%, administrada durante 12-14 horas en infusión
continua. Debido a que requiere hospitalización, se recomienda 1-2 días cada 3-4
semanas y las siguientes indicaciones: disminuir rápidamente la sobrecarga de hierro
previo al trasplante de células progenitoras hematopoyéticas, posterior a la transfusión

62 de concentrado eritrocitario y previo al inicio de quelante oral. No se recomienda la
administración intramuscular por su baja acción quelante.
Como efectos colaterales se pueden presentar reacciones locales en el sitio de
aplicación, así como oftálmicas, auditivas y óseas. Existen reportes sobre la toxicidad
pulmonar aguda con hipoxemia y fibrosis intersticial en pacientes que han sido tratados
a largo plazo con dosis altas deeste quelante.
Deferiprona. Fue el primer desarrollo de un quelante oral. Se trata de un quelante
bidentado en el que tres moléculas del mismo se unen por cada molécula de hierro. Se
absorbe rápidamente y alcanza concentraciones pico en 45-60 min. Su vida media
plasmática ha sido estimada en 91.1 min22 y se inactiva en el hígado por
glucouridación, lo que induce la excreción de hierro casi exclusivamente en la orina y
sólo una pequeña parte en heces. La dosis usual es de 75 mg/kg/día.
Existen pocos estudios que comparan a la deferiprona con la deferoxamina. Un
trabajo multicéntrico, prospectivo y aleatorizado en 144 pacientes talasémicos con
valores basales de ferritina sérica de hasta 3 000 μg/L recibieron deferiprona a 75
mg/kg/día en tres tomas, o bien, deferoxamina en dosis de 50 mg/kg/día subcutánea a
lo largo de 12 horas, cinco días a la semana por un año.24 No se registraron diferencias
significativas en la tasa de reducción promedio de ferritina sérica: 222 μg/L y 232 μg/L
para deferiprona y deferoxamina, respectivamente.
Por último, también se ha reportado que deferoxamina es inferior a deferiprona en
función de la protección cardiovascular. Los efectos adversos más comunes de la
deferiprona consisten en cambios de color en la orina y molestias a nivel
gastrointestinal (náusea, vómito, dolor abdominal) que suelen ser controlables. La
agranulocitosis es el efecto adverso más severo, pero se desconoce el mecanismo que
la desencadena; su incidencia es baja y remite al descontinuar el tratamiento.
Deferasirox. Es el quelante de más reciente desarrollo, diseñado con un modelo
molecular que forma parte de la familia conocida como bi-hidroxi-fenil-triazoles. Se une
al hierro en una relación de 2:1, se absorbe rápidamente, alcanza niveles plasmáticos

63 pico en 1-3 horas, es altamente selectivo para este mineral, sin afectar los niveles de
zinc y cobre, 26 y la excreción del hierro es por heces. Su vida media en plasma es de
11-19 horas y se puede prolongar empleando dosis altas. Se ha demostrado que con
dosis de 10-40 mg/kg se puede conseguir una tasa de excreción de hierro de 0.3
mg/kg/día, la cual puede ser suficiente para mantener el equilibrio férrico en pacientes
bajo terapia con HT.
Este fármaco posee una potencia comparable a la deferoxamina, es seguro y de
fácil administración por vía oral. Para una mejor absorción debe tomarse 30 minutos
antes del desayuno, disuelto en jugo de manzana o naranja, evitando mezclarlo con
utensilios. Entre los efectos adversos se presentan cefalea, erupción cutánea, efectos
gastrointestinales como náusea, vómito y dolor abdominal que no requieren o raras
veces necesitan ajuste de dosis. Su utilización requiere el control periódico de la función
renal y hepática y la valoración oftalmológica y auditiva anual con potenciales
evocados.
Es importante identificar altos requerimientos de HT en el paciente, el momento en
que se presenta la sobrecarga de hierro e iniciar oportunamente el tratamiento de
quelación. Posteriormente, es necesario el control estricto y la vigilancia de la respuesta
al tratamiento, la identificación de los efectos no deseados de las drogas quelantes y el
momento en que se debe descontinuar éstas al tener niveles de ferritina sérica <500
μg/L.
En el paciente que ha llegado al equilibrio de hierro, pero que requiere continuar
con la administración de concentrado eritrocitario periódicamente, deberá realizarse una
monitorización de la ferritina sérica trimestralmente y evaluar el uso de quelantes de
hierro si los requerimientos transfusionales son ≥120 mL/kg/día.
MARCO TEÓRICO OPERACIONAL
2.3 Hipótesis:

64 Los pacientes con AF y Talasemia muestran valores elevados de hierro orgánico
con riesgo elevado de desarrollar hemocromatosis, motivado al régimen transfusional al
cual son sometidos.
2.4 Variables: Hemoglobinopatías
Definición Conceptual: son alteraciones cualitativas o cuantitativas de la globina,
secundarias a mutaciones genéticas, cuya consecuencia puede ser una modificación
estructural (hemoglobinopatías estructurales entre ellas la Anemia Falciforme) o una
disminución de la síntesis de una cadena de globina estructuralmente normal
(talasemias). (Malcorra JJ. 2001; Fairbanks VF, 1980)
Concentraciones Fe de hierro sérico, TIBC, %ST, Ferritina sérica: parámetros
bioquímicos que permiten conocer el estado del hierro orgánico y permiten el
diagnóstico de ferropenia o hemosiderosis (Pérez Surribas y Viedma JA, 2006) Definición Operacional:
Hemoglobinopatías enfermedades genéticas con alteración de la hemoglobina que
conducen a anemia crónica y ameritan de transfusión frecuentemente con el riesgo de
producir sobrecarga de hierro en sangre.
Concentraciones Fe de hierro sérico, TIBC, %ST, Ferritina sérica: parámetros
bioquímicos que miden el hierro orgánico y permiten el diagnóstico de sobrecarga de
hierro o hemosiderosis.
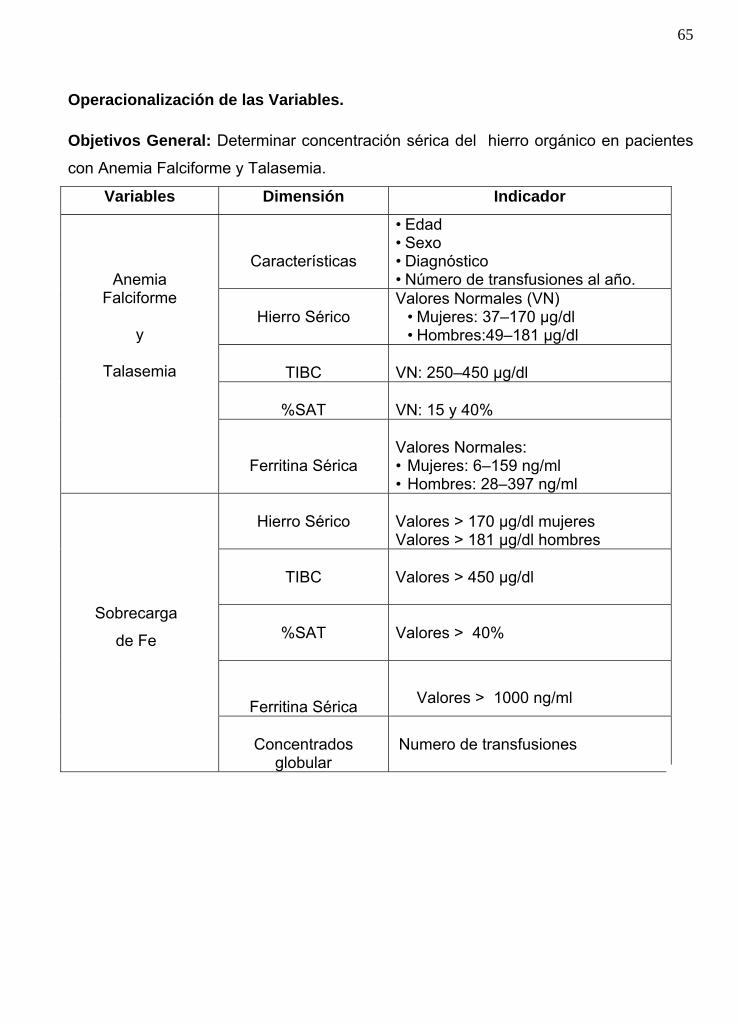
65 Operacionalización de las Variables.
Objetivos General: Determinar concentración sérica del hierro orgánico en pacientes
con Anemia Falciforme y Talasemia.
Variables Dimensión Indicador
Anemia Falciforme
y
Talasemia
Características
• Edad • Sexo • Diagnóstico • Número de transfusiones al año.
Hierro Sérico
Valores Normales (VN) • Mujeres: 37–170 µg/dl • Hombres:49–181 µg/dl
TIBC
VN: 250–450 µg/dl
%SAT
VN: 15 y 40%
Ferritina Sérica
Valores Normales: • Mujeres: 6–159 ng/ml • Hombres: 28–397 ng/ml
Sobrecarga
de Fe
Hierro Sérico
Valores > 170 µg/dl mujeres Valores > 181 µg/dl hombres
TIBC
Valores > 450 µg/dl
%SAT
Valores > 40%
Ferritina Sérica
Valores > 1000 ng/ml
Concentrados
globular Numero de transfusiones

66
CAPITULO III
MARCO METODOLOGICO

67 3.1. TIPO DE INVESTIGACIÓN La investigación se tipifica de la siguiente manera:
Investigación descriptiva y correlacional Según Ramírez (1999), En este tipo de investigación se busca la descripción, con
mayor precisión de las características de un determinado individuo, situaciones o
grupos y cuyo alcance determina la frecuencia con la que algo ocurre o con la que algo
se halla asociado con otro factor. De acuerdo con lo anterior, este estudio es de tipo descriptivo, debido a que se
busca evaluar la utilidad de los niveles de hierro orgánico como pronostico en
hemosiderosis en los pacientes con Anemia Falciforme y Talasemia.
Será un estudio correlacional, los cuales según Hernández y col (1998; 62) ya que se
van a establecer relaciones que responden a las causas de los eventos físicos o
sociales”.
Investigación transversal. El presente estudio es una investigación transversal, debido a que los criterios de
los grupos son medidos en un momento, sin evaluar la evolución de estos en el tiempo.
(Chávez, 1994)
3.2. DISEÑO DE LA INVESTIGACIÓN El termino diseño se refiere al plan o estrategia concebida para responder a las
preguntas de investigación (Christensen 1980); y señala al investigador lo que debe
hacer para alcanzar su objetivo de estudio y contestar interrogantes que se ha
planteado.
El diseño de la investigación fue no experimental ya que se limita a la observación
de las diversas respuestas manifestadas por los elementos en estudio ante situaciones
que no son manipuladas por el investigador o en las que la ubicación de los elementos
no depende del investigador, en este caso se evaluó la utilidad de los niveles de hierro
orgánico como pronostico en hemosiderosis en los pacientes con Anemia Falciforme y
Talasemia atendidos en el Servicio de Hematología del Hospital Central Dr Urquinaona
y el Instituto Hematológico de Occidente Banco de Sangre del estado Zulia.

68
3.3. MATERIALES Y MÉTODO 3.3.1. Población: La población de esta investigación estuvo formada por todos los pacientes con
diagnóstico de Anemia Falciforme y talasemia, de ambos sexos y de cualquier edad
que acudieron al Servicio de Hematología del Hospital Central Dr. Urquinaona y el
Instituto Hematológico de Occidente Banco de Sangre del estado Zulia.
. 3.3.2. Muestra
La muestra de tipo no probabilística según Parra (2000), es aquella en la que la
elección se realiza a través factores diferentes al azar, es por esto que la muestra en la
presente investigación es no probabilística debido a que se seleccionaron todos los
pacientes con diagnostico establecido de AF y Talasemia de ambos sexos y de
cualquier edad que se estudiaron desde Marzo 2009 a Julio 2010, la cual fue de 33
pacientes.
3.3.3. Criterios de Inclusión y Exclusión:
Se incluyeron pacientes de cualquier edad, ambos sexo, con diagnóstico de AF
y Talasemia que acudieron a la consulta del Servicio de hematología del hospital Dr
Urquinaona y del Banco de sangre de occidente durante el periodo estudiado.
Se excluyeron los pacientes de cualquier edad y sexo con diagnóstico de AF y
Talasemia que hayan fallecido o que ya no vivan en el estado.
3.4. Método
Previa autorización del paciente se realizó examen físico, seguidamente se
tomaron muestra sanguínea, específicamente 10 ml de sangre venosa, en ayunas, la
cual se depositó en tubos estandarizados, limpios de vidrio, posteriormente se
centrifugaron a alta revolución durante 5 minutos para obtener el suero, el cual se
conservó por 3 días entre 2-8 ºC o durante 6 meses a -20 ºC.

69 Para realizar este procedimiento se necesitó analizador espectrofotómetro o
fotómetro y Kit hierro-ferrozina. Para realizar los cálculos de capacidad total de fijación
de hierro (TIBC), se utilizó la fórmula de TIBC= concentración de hierro en el
sobrenadante de la muestra centrifugada x 3 (diluciones) y para la saturación de hierro
(SAT%) 100 x concentración de hierro sérico/TIBC= SAT%.
3.5. Técnica de Recolección de Datos
Para Sabino (1992), un instrumento de recolección de datos es cualquier recurso
del que se vale el investigador para acercarse al fenómeno estudiado y obtener
información de él, además de considerarse el elemento vinculante entre la teoría y los
hechos.
Dentro de la gran diversidad de instrumentos de recolección de datos, existen
ciertas técnicas que son más utilizadas de acuerdo al tipo de investigación realizada.
Se realizo un cuestionario de recolección de datos diseñado por el investigador,
donde se recolectaron datos personales, diagnóstico, tratamiento transfusional y
quelante recibido, fecha, parámetros de laboratorio a la que se le aplicara validez y
confiabilidad de contenido a través de juicio de expertos.
3.6. Técnica de Análisis de Datos Esta se realizo en dos etapas, una primera etapa es la tabulación de los datos y una
segunda etapa que se refiere al análisis de los datos.
La tabulación de los resultados se realiza en dos procesos según sabino (1991), el
primero es el conteo de las observaciones de cada una de las categorías, el segundo
es la presentación de los resultados ordenados en tablas.
En este estudio los datos son presentados como frecuencias absolutas y relativas.
Cuyas definiciones según Hernández, Fernández y Baptista (1988), son, frecuencia
absoluta es el registro del número de veces que se repita un dato, mientras que las
frecuencias relativas son su expresión porcentual.
Según Chávez (2001), el análisis de los datos se deriva de las frecuentes
comparaciones que se producen entre los resultados de mayor y menor puntaje.

70 En el mismo tema, Sabino (1996), plantea que el análisis descriptivo identifica las
características más resaltantes de las variables estudiadas en una investigación.
Tomando lo anteriormente expuesto se infiere que el análisis descriptivo de cada
una de las variables fue el más apropiado para evaluar los resultados de esta
investigación.

71
CAPITULO IV
ANALISIS Y DISCUSION DE LOS RESULTADOS

72 4.1 RESULTADOS
Tabla 1. CARACTERÍSTICAS GENERALES DE LOS PACIENTES CON ANEMIA
FALCIFORME Y TALASEMIA ESTUDIADOS
Edad en años (Promedio ± Desviación estándar, Rango)
38,08 2 ± 12,96
(Rango= 21-61)
Sexo Masculino (M) y Femenino (F) M: 12/26 (46,15%)
F: 14/26 (53,85%)
No de casos con Anemia Falciforme 18/26 (69,24%)
F: 10/18 (55,5%)
M: 8/18 (44,4%)
No de casos con Talasemias 8/26 (30,76%)
F: 4/8 (50%)
M: 4/8 (50%)
Promedio de transfusiones en el último año 11,92 ± 8,22
Rango: (0-24).
No: Número de casos Fe: Hierro
En la Tabla 1 muestra las características generales de los pacientes con Anemia
Falciforme y Talasemia estudiados, se puede evidenciar que la edad promedio fue de
38.08 ± 12,96 años (rango 21-61 años), predominando el sexo femenino con 53,85%. El
69,24% correspondió a Anemia Falciforme el resto lo conformaron pacientes con
diagnóstico de Talasemia. El promedio de transfusiones de concentrados eritrocitarios
anual fue de 11,92 ± 8,22 (rango: 0-24), estos datos revelan que la AF y la talasemia
puede manifestarse a cualquier edad, con una mayor frecuencia para el sexo femenino
y mayor frecuencia para AF con respecto a talasemia en una proporción 3:1
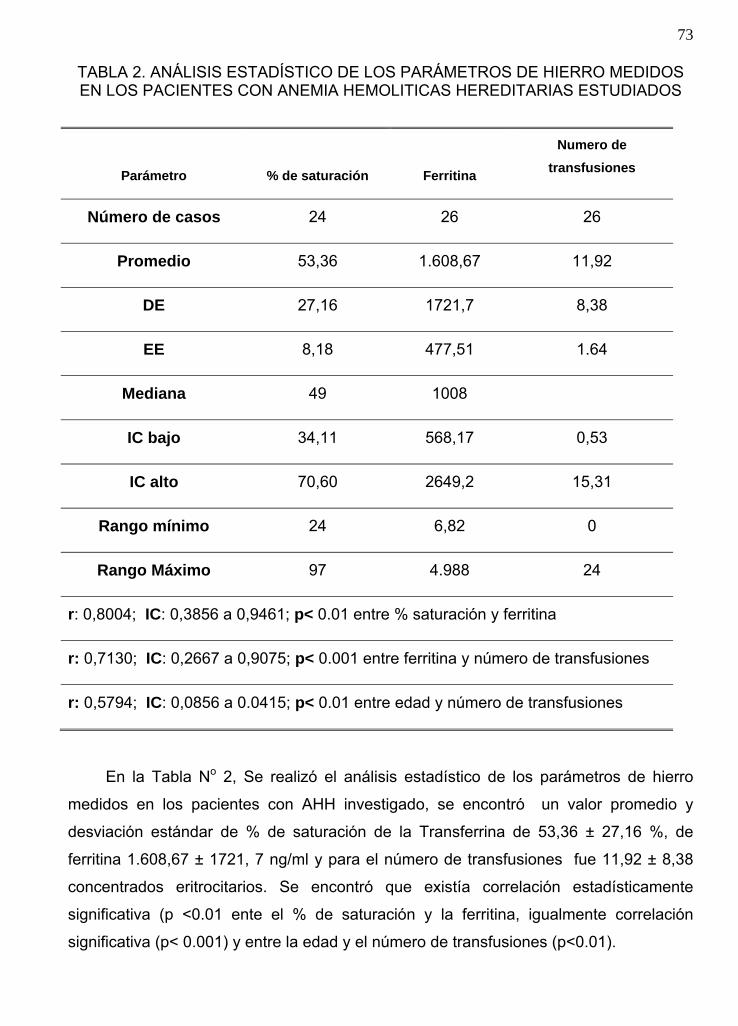
73
TABLA 2. ANÁLISIS ESTADÍSTICO DE LOS PARÁMETROS DE HIERRO MEDIDOS EN LOS PACIENTES CON ANEMIA HEMOLITICAS HEREDITARIAS ESTUDIADOS
Parámetro
% de saturación
Ferritina
Numero de transfusiones
Número de casos 24 26 26
Promedio 53,36 1.608,67 11,92
DE 27,16 1721,7 8,38
EE 8,18 477,51 1.64
Mediana 49 1008
IC bajo 34,11 568,17 0,53
IC alto 70,60 2649,2 15,31
Rango mínimo 24 6,82 0
Rango Máximo 97 4.988 24
r: 0,8004; IC: 0,3856 a 0,9461; p< 0.01 entre % saturación y ferritina
r: 0,7130; IC: 0,2667 a 0,9075; p< 0.001 entre ferritina y número de transfusiones
r: 0,5794; IC: 0,0856 a 0.0415; p< 0.01 entre edad y número de transfusiones
En la Tabla No 2, Se realizó el análisis estadístico de los parámetros de hierro
medidos en los pacientes con AHH investigado, se encontró un valor promedio y
desviación estándar de % de saturación de la Transferrina de 53,36 ± 27,16 %, de
ferritina 1.608,67 ± 1721, 7 ng/ml y para el número de transfusiones fue 11,92 ± 8,38
concentrados eritrocitarios. Se encontró que existía correlación estadísticamente
significativa (p <0.01 ente el % de saturación y la ferritina, igualmente correlación
significativa (p< 0.001) y entre la edad y el número de transfusiones (p<0.01).
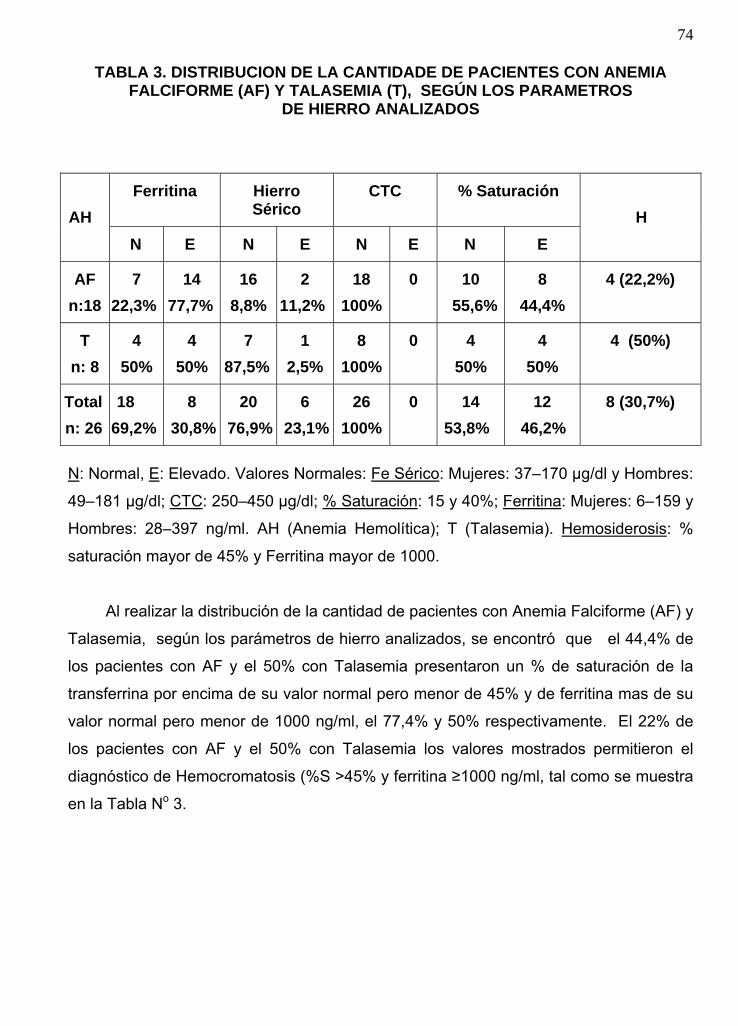
74
TABLA 3. DISTRIBUCION DE LA CANTIDADE DE PACIENTES CON ANEMIA FALCIFORME (AF) Y TALASEMIA (T), SEGÚN LOS PARAMETROS
DE HIERRO ANALIZADOS
AH
Ferritina Hierro Sérico
CTC % Saturación H
N E N E N E N E
AF n:18
7 22,3%
14 77,7%
16 8,8%
2 11,2%
18 100%
0 10 55,6%
8 44,4%
4 (22,2%)
T n: 8
4 50%
4 50%
7 87,5%
1 2,5%
8 100%
0 4 50%
4 50%
4 (50%)
Total n: 26
18 69,2%
8 30,8%
20 76,9%
6 23,1%
26 100%
0 14 53,8%
12 46,2%
8 (30,7%)
N: Normal, E: Elevado. Valores Normales: Fe Sérico: Mujeres: 37–170 µg/dl y Hombres:
49–181 µg/dl; CTC: 250–450 µg/dl; % Saturación: 15 y 40%; Ferritina: Mujeres: 6–159 y
Hombres: 28–397 ng/ml. AH (Anemia Hemolítica); T (Talasemia). Hemosiderosis: %
saturación mayor de 45% y Ferritina mayor de 1000.
Al realizar la distribución de la cantidad de pacientes con Anemia Falciforme (AF) y
Talasemia, según los parámetros de hierro analizados, se encontró que el 44,4% de
los pacientes con AF y el 50% con Talasemia presentaron un % de saturación de la
transferrina por encima de su valor normal pero menor de 45% y de ferritina mas de su
valor normal pero menor de 1000 ng/ml, el 77,4% y 50% respectivamente. El 22% de
los pacientes con AF y el 50% con Talasemia los valores mostrados permitieron el
diagnóstico de Hemocromatosis (%S >45% y ferritina ≥1000 ng/ml, tal como se muestra
en la Tabla No 3.
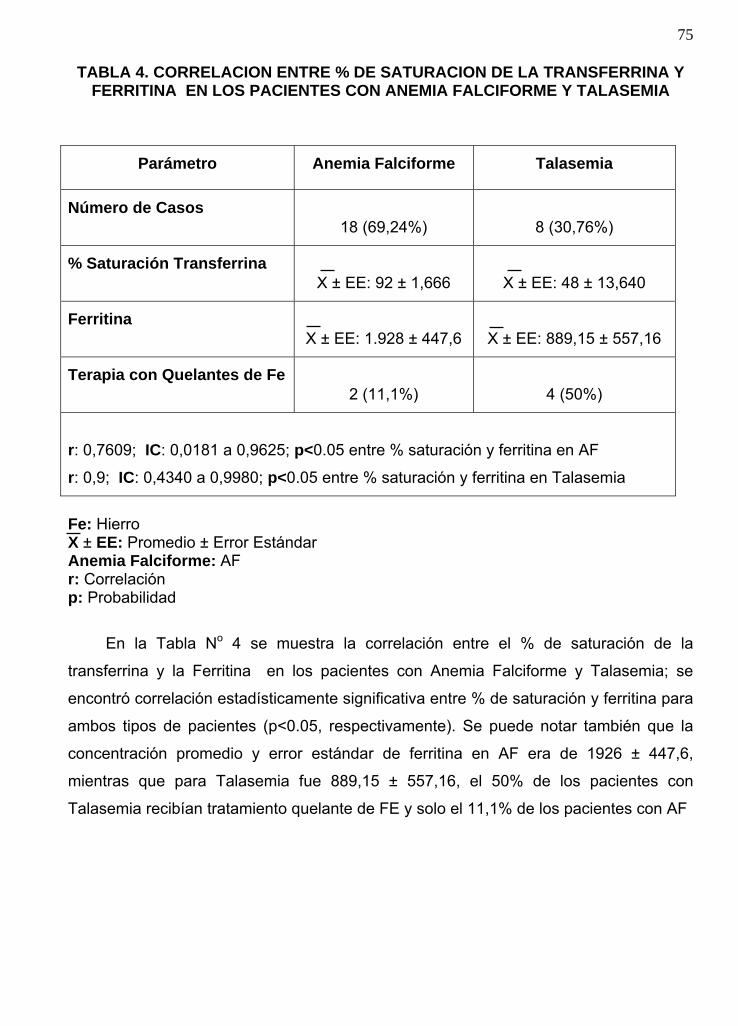
75
TABLA 4. CORRELACION ENTRE % DE SATURACION DE LA TRANSFERRINA Y FERRITINA EN LOS PACIENTES CON ANEMIA FALCIFORME Y TALASEMIA
Parámetro Anemia Falciforme Talasemia
Número de Casos
18 (69,24%)
8 (30,76%)
% Saturación Transferrina
X ± EE: 92 ± 1,666
X ± EE: 48 ± 13,640
Ferritina
X ± EE: 1.928 ± 447,6
X ± EE: 889,15 ± 557,16
Terapia con Quelantes de Fe
2 (11,1%)
4 (50%)
r: 0,7609; IC: 0,0181 a 0,9625; p<0.05 entre % saturación y ferritina en AF
r: 0,9; IC: 0,4340 a 0,9980; p<0.05 entre % saturación y ferritina en Talasemia
Fe: Hierro X ± EE: Promedio ± Error Estándar Anemia Falciforme: AF r: Correlación p: Probabilidad
En la Tabla No 4 se muestra la correlación entre el % de saturación de la
transferrina y la Ferritina en los pacientes con Anemia Falciforme y Talasemia; se
encontró correlación estadísticamente significativa entre % de saturación y ferritina para
ambos tipos de pacientes (p<0.05, respectivamente). Se puede notar también que la
concentración promedio y error estándar de ferritina en AF era de 1926 ± 447,6,
mientras que para Talasemia fue 889,15 ± 557,16, el 50% de los pacientes con
Talasemia recibían tratamiento quelante de FE y solo el 11,1% de los pacientes con AF

76
DISCUSION
La revisión de la literatura sobre la epidemiología de la sobrecarga de hierro en
pacientes con Anemias Hemolíticas Hereditaria en América Latina, son escasos. Debido
a esta situación, se inició un estudio denominado “Registro de Pacientes en América
Latina con Hemosiderosis transfusional” (RELATH) (Araújo, 2009) en los cuales
participaban nueve países a saber: Argentina, Brasil, Colombia, Ecuador, México,
Panamá, Perú, Trinidad y Tobago y Venezuela. Los resultados preliminares de este
estudio incluyeron los datos de 236 pacientes en total. En el presente trabajo el número
de casos estudiados fue mucho menor que el reportado por RELATH, el total fue de
solo 26, este es debido a varias razones: (1) Solo corresponden a pacientes con
Anemias Hemolíticas Hereditarias como AF y Talasemias. (2) Los pacientes se ubican
solo en un estado de Venezuela. (3) Los pacientes incluidos únicamente correspondían
a dos centros asistenciales, siendo estos el lugar donde se realiza el postgrado
universitario de Hematología del estado Zulia. (4) El número de casos que se planificó
estudiar eran pocos dado el costo que representaba los estudios de laboratorios que
debían practicarse.
De igual manera, en el estudio RELATH (Araújo, 2009) los resultados muestran
que los pacientes investigados tenían una edad media de 29 ± 20.4 años, más baja que
la edad promedio de los pacientes estudiados en el presente trabajo la cual se
encontraba en 38,08 2 ± 12,96 (rango: 21-61); mientras que el sexo predominante fue el
femenino, en el RELATH fue de 54% y en el nuestro de 53,85%.
En el mismo estudio, se señalan como los diagnósticos más frecuentes: Anemia
Falciformes en el 44%, 17% para Talasemia, 14% en Anemia Aplásica y Síndromes
Mielodisplásicos 7% (Araújo, 2009). En este trabajo el 69,24% correspondió Anemia
Falciforme y el 30,76% pata Talasemia. Al respecto, en Venezuela la frecuencia para
hemoglobinopatías se indica en 9%, siendo la Drepanocitosis la más frecuente con
69,8% (Arends y col, 2007), cifras que oscilan entre 0-7% dependiendo del componente
africano de la región (Salazar-Lugo, 2004); así en estado Zulia, se reporta para Isla de
Toas un 13% (Pineda-Del Villar y Borjas M, 1986) y para las regiones aledañas 6,4%
(Torres-Guerra y col, 1993), en ambas zonas la característica fenotípicas de su

77 población es caucásica. Mientras que un menor porcentaje se le reconoce a las
Talasemia a las cuales se les observa en 2% (Arends y col, 2007).
El número promedio anual de transfusiones sanguíneas recibidas por los
pacientes con Anemias Hemolíticas Hereditarias estudiadas fue de 11,92 ± 8,22 (rango:
0-24) en el último año, similar a lo observado en otros estudios en los cuales se
describe en 12 ± 8.8 anualmente (Araújo, 2009). Al respecto, se indica que al
transfundir una cantidad igual o mayor de 120 mL/kg de concentrados de glóbulos rojos
(U.S. Department of Health and Human Services, 2004) se produce sobrecarga. Esta
sobrecarga puede detectarse después de las primeras 10-20 transfusiones, lo que
ocurre aproximadamente cerca de los 3 años de iniciar las transfusiones (Cappellini y
col., 2000), siendo este el riego que tienen los pacientes del presente estudio, dado la
edad que presentan y el número de transfusiones recibidos anualmente.
El diagnóstico de sobrecarga de hierro esta basado en la sospecha clínica y la
medición del hierro sérico, que no tiene valor por sí solo y además debe tenerse ciertas
precauciones al momento de su determinación ya que se encuentra influido por el ciclo
circadiano, ingestión de alimentos y enfermedades inflamatorias; situación que también
se presenta para la ferritina que aumenta en procesos inflamatorios, infección,
hepatopatías, entre otras patologías. Por ello se hace necesario conocer además el
porcentaje de saturación de transferrina que se encontrará completamente saturada y
se correlaciona bien con la Ferritina (Borgna-Pignatti y Galanello, 2003). Pero el %ST
se eleva más precozmente que la Ferritina plasmática y se sospecha de sobrecarga
cuando su valor es mayor de 45%. El diagnóstico de sobrecarga de hierro se establece
al obtener un %ST mayor de 45% (mayor del 50% en mujeres y 60% en varones) y una
Ferritina mayor de 1000 ng/ml (Powell y col, 2004; Bacon y col, 1996; Ladero y col,
2005; Roa y col, 2001).
En el presente estudio se encontró un 23,1% de pacientes con AHH con exceso
de Fe sérico (11,2% con AF y 2,5% para Talasemia), el valor promedio fue 136,27 ±
57,376 µg/dl. El %ST promedio fue 53,363 ± 27,16%, estuvo elevado en 46,2% de los
pacientes (44,4% en AF y 50% en Talasemia). Para la ferritina el promedio fue 1.608,67
± 1721,7 ng/ml y estuvo elevada en 30,8% de los pacientes (77,7% en AF y 50% para

78 Talasemia) y se encontró Hemocromatosis en 30,7% (22,2% en AF y 50% en
Talasemia) considerándose para valores >45% para ST y >1000 ng/mL para ferritina.
Sin embargo, se reporta que un valor de Ferritina sérica >200 mcg% en mujeres pre-
menopáusicas, y 300 mcg en el varón y en la mujer post-menopáusica indica
sobrecarga pero no hemocromatosis, al igual manera que un valor >450 mcg de
Ferritina sérica (Remacha y col., 2000); otros describen que un %ST >55% con Ferritina
normal indica hemocromatosis aun no expresada y puede existir hiperferritinemia con
sobrecarga de hierro y saturación de transferrina normal (Moirand y col., 1997). La
sideremia por ser es muy cambiante tiene poco valor (Powell y col., 1998).
Nuestros resultados muestran un valor promedio de ferritina de 1.608,67 ± 1721,7
ng/ml, mucho mas bajo que el descrito por Taher A, y col. (2001), quienes encontraron
en 56 pacientes con Talasemia del Líbano, una concentración de Ferritina sérica de
3.663+/-566 microg/l y que el reportado por el RELATH cuyo nivel medio de Ferritina
sérica fue 2.564 ± 1.834 mg/l. Tanto en el caso de los trabajos de los autores
contrastados como en el presente trabajo, las evaluaciones para medir la concentración
hepática de hierro (LIC) no se realizaron y la sobrecarga de hierro solo se evaluó
usando los niveles de Ferritina sérica y el % de Saturación de la Transferrina. Al
contrario de estudio de Brown y col., quienes estudiaron pacientes con AF y
determinaron hierro hepático obtenido por biopsia, concentración de Ferritina y
Saturación de la Transferrina y encontraron valores altos de Fe en hígado, Ferritina
elevada y %ST mayor de 45%, con una correlación significativa entre estos parámetros,
por lo que sugieren que el estudio de tejido es importante de considerar a la hora de
realizar el diagnóstico definitivo de sobrecarga de Fe y el cual se considera de realizar
en la presente investigación.
De igual forma, Koren y col., estudiaron pacientes con AF y Talasemia en quienes
midieron Fe no unido a la transferrina (HNUT) y Fe plasmático lábil (HPL). Señalan que
ningún paciente con AF resultó positivo para HNUT, pero si 17/43 con Talasemia. 4
pacientes con Talasemia y 1 de AF resultaron positivos para HPL, estos resultados les
permite concluir que pacientes con AF aun cuando son politransfundidos, presentan un
comportamiento diferente que los pacientes con Talasemia y estos son los que tienen
mayor riesgo de desarrollar sobrecarga de Fe. Este hallazgo también lo describe

79 Vázquez y col (2002), quienes encontraron que la sobrecarga de hierro inducida por
transfusiones es la consecuencia fatal mas frecuente en los pacientes que recibieron
con la transfusión crónica para diferentes tipos de anemias, pero que los niños con
talasemia constituyeron el grupo más grandemente afectado.
Lo descrito en el párrafo anterior se refleja en los resultados obtenidos en el
presente estudio, pues el número de casos con hemocromatosis fue del 50% para
Talasemia y solo el 22,55 para AF, a pesar de que el 50% de los pacientes con
Talasemia recibían tratamiento con quelantes de Fe y solo el 11,1 % de aquellos con
AF, haciéndolos mas susceptibles de presentar sobrecarga de Fe.
En ese sentido se establece que los niveles de Ferritina sérica y de Saturación de
Tansferrina deben ser los métodos de primera línea para el diagnóstico y monitoreo de
la sobrecarga de hierro, la primera deberá determinarse cuando se realiza el
diagnóstico y cuantificarse mensualmente hasta que se haya establecido una
tendencia. Posteriormente, se deberá evaluar al paciente cada 3 meses. Existe la
necesidad de desarrollar otros métodos cuantitativos para obtener valores mas precisos
como la Resonancia Magnética (RM) para el diagnóstico y el monitoreo de
hemosiderosis hepática, acompañada, si es posible de biopsia hepática para confirmar
el diagnóstico y confirmar los resultados de la RM. De igual manera es importante
considerar la realización de ecocardiografía Doppler bidimensional para diagnóstico y
monitoreo de hemosiderosis cardíaca, y en lo posible acompañarse con una RM
cardíaca (Cappellini y col., 2000).
Es importante destacar que la Federación Internacional de Talasemia (Cappellini y
col., 2000) enfatiza que siendo la sobrecarga de hierro la complicación más importante
en las AHH debe ser el principal foco de atención para el tratamiento clínico de esta
complicación, por ello se han propuesto pautas para su control, entre estas se indican:
• Exámenes de detección de sobrecarga al comienzo de las transfusiones.
• Controlar la concentración de ferritina sérica por lo menos cada 3 meses, pero no
confiar en esta determinación solamente,
• Conocer la concentración de hierro hepático, que se determina a través de
biopsia o de forma no invasiva por Resonancia Magnética, recientemente se sugiere la
realización de la susceptometría biomagnética (SQUID) estos exámenes se consideran

80 como el estándar de referencia para calcular la carga de hierro pero no se dispone en la
mayoría de los países del mundo y su costo es muy elevado.
Una vez que se haga el diagnóstico de sobrecarga de Fe, deberá iniciarse el
tratamiento con agentes quelantes del hierro, bien cuando la Ferritina sérica este por
encima de 1000 μg/l o después de que el paciente haya recibido transfusión unidades
de glóbulos rojos entre 10 a 20, independiente del tiempo. Este tratamiento debe
acompañarse de la vigilancia de las pruebas hepáticas y renales mensualmente
(alanina-aminotransferasa, aspartato aminotransferasa, creatinina sérica, proteinuria),
seguimiento de la función cardíaca cada 6 meses con una ecocardiografía Doppler
bidimensional y anualmente los pacientes deben someter a evaluaciones oftalmológicas
y otolaringológicas (Cappellini y col., 2000).
En resumen, la determinación de los parámetros de Fe en los pacientes con AF y
Talasemia estudiados, permiten identificar aquellos pacientes con sobrecarga de Fe o
Hemocromatosis, para iniciar estudio de los diferentes órganos y conocer si existe
anormalidad o no. También se deberá indicar tratamiento con agentes quelantes de
Hierro, el cual se distribuye en nuestro país de manera gratuita. Este tratamiento deberá
contar con una vigilancia clínica y de laboratorio estricta, a fin de conocer la evolución
de la sobrecarga de FE en el paciente, así como también identificar a tiempo sus
efectos indeseables.

81
CAPITULO V
CONCLUSIONES Y RECOMENDACIONES

82
CONCLUSIONES
1. La edad promedio de los pacientes con AHH estudiados fue 38.08 ± 12,96 años
(rango 21-61 años)
2. El sexo que predominó fue el femenino con 53,85%.
3. El 69,24% de los casos estudiados correspondió a Anemia Falciforme el resto lo
conformaron pacientes con diagnóstico de Talasemia.
4. El promedio de transfusiones de concentrados eritrocitarios anual fue de 11,92 ± 8,22
(rango: 0-24).
5. El 44,4% de los pacientes con AF y el 50% con Talasemia presentaron un % de
saturación de la transferrina por encima de su valor normal y de ferritina mayor de su
valor normal, el 77,4% y 50% respectivamente.
6. El 22% de los pacientes con AF y el 50% con Talasemia, mostraron valores que
permiten el diagnóstico de Hemocromatosis (%S >45% y ferritina ≥1000 ng/ml).
7. El valor promedio y desviación estándar del % de saturación de la Transferrina fue de
53,36 ± 27,16 %, e ferritina 1.608,67 ± 1721, 7 ng/ml y el número de transfusiones
fue 11,92 ± 8,38 de concentrados eritrocitarios.
8. Se encontró correlación estadísticamente significativa (p <0.01) ente el % de
saturación y la Ferritina, igualmente se observó correlación significativa (p< 0.001) y
entre la edad y el número de transfusiones (p<0.01) en los pacientes con AHH. 9. Al discriminar según el diagnóstico de AHH estudiado se encontró correlación entre
el % de saturación de la transferrina y la Ferritina en los pacientes con Anemia
Falciforme y Talasemia; se encontró correlación estadísticamente significativa entre
% de saturación y ferritina para ambos tipos de pacientes (p<0.05, respectivamente). 10. La concentración promedio y error estándar de la Ferritina en AF era de 1926 ±
447,6, mientras que para Talasemia fue 889,15 ± 557,16 11. El 50% de los pacientes con Talasemia recibían tratamiento quelante de FE y solo
el 11,1% de los pacientes con AF

83
RECOMENDACIONES
1. Se recomienda seguir las Pautas de la Federación Internacional de Talasemia para
los pacientes con AHH, a saber:
• Exámenes de detección de sobrecarga al comienzo de las transfusiones.
• Controlar la concentración de ferritina sérica por lo menos cada 3 meses.
• Conocer la concentración de hierro hepático, a través de biopsia o por
Resonancia Magnética
• Estudio de función cardiaca.
2. En aquellos casos en los cuales se diagnostique sobrecarga de hierro, indicar
terapia con quelantes de Fe 3. Se necesita controlar a los pacientes que reciben terapia con agentes a través de
pruebas hepáticas y renales mensualmente, seguimiento de la función cardíaca
cada 6 meses y anualmente evaluaciones oftalmológicas y ORL. 4. En los pacientes estudiados que presentaron sobrecarga de hierro y no reciben
terapia con agentes quelantes administrar esta y evaluar tejido hepático y cardiaco.

84
LITERATURA CITADA

85
REFERENCIAS BIBLIOGRAFICAS
1. Altes A, Remacha AF, Sureda A, Martino R, Briones J, Brunet S, et al. Patients
with biochemical iron overload: causes and characteristics of a cohort of 150 cases. Ann Hematol 2003; 82: 127-30.
2. Altes A. Sobrecarga férrica. Algo más que hemocromatosis hereditaria. Med Clin (Barc) 2003; 120:704-6.
3. Altésa M A, Conxa Castell AR, Roure E, Tresserras R. Déficit y sobrecarga de hierro en la población adulta de Cataluña. Disponible en http://www.doyma. 14/02/2007.
4. Araújo A. Fundação Hemope. V Simposio Basileiro Doença Falciforme e outras hemoglobinopatías. Encontro Panamericano para doenca falciforme OPAS/OMS, Recife-Brasil. 2009. Disponible en www.cehmob.org.br/simposio/pdf. 20/05/2011)
5. Arends A, Chacín M, Bravo-Urquiola M, Montilla S, Guevara JM, Velazquez D, García G, Alvarez M, Castillo O (2007). Hemoglobinopatía en Venezuela.Interciencia;31 (8): 516-521.
6. Bacon BR, Tavill AS (1996). Hemohromatosis and the iron overload syndromes. En: Zakim D, Boyer TD. Editores. Hepatology: a textbook of liver disease. Filadelfia: WB Saunders; 1.439- 1.472.
7. Borgna-Pignatti C, Galanello R. Thalassemias and related disorders: quantitative disorders of hemoglobin synthesis. Wintrobe’s Clinical Hematology. Philadelphia: Lippincott Williams & Wilkins; 2003. p. 1319-1365)
8. Brown K, Subramony C, May W, Megason G, Liu H, Bishop P, Walker T, Nowicki MJ. Hepatic iron overload in children with sickle cell anemia on chronic transfusion therapy. J Pediatr Hematol Oncol. 2009 May;31(5):309-12
9. Bunn HF and Forget BG. 1986. Hemoglobin: Molecular, Genetics, and Clinical Aspects. W.B. Saunders.
10. Cano-Castellanos R, López-Santiago N, Piedras J. 2009. Sobrecarga de hierro en pacientes pediátricos. Bol Med Hosp Infant Mex 66, noviembre-diciembre.
11. Carrondo MA. Ferritins, iron uptake and storage from the bacterioferritin viewpoint. EMBO J 2003; 22: 1959-68.
12. Cappellini N, Cohen A, Eleftheriou A, Piga A, Porter J. Iron Overload. En: Guidelines for the Clinical Management of Thalassaemia: Thalassaemia International Federation; Capítulo 5, 2000:21-35
13. Cappellini N, Cohen A, Eleftheriou A, Piga A, Porter J. Iron Overload. En: Guidelines for the Clinical Management of Thalassaemia. Thalassaemia International Federation; Capítulo 2. 2000: 5-7
14. Gabutti V, Borgna_Pignatti C. 1994. Clinical manifestations and therapy of transfusional Haemosiderosis. Baillieres Clin Haematol 7: 919-940
15. García Rodriguez C, Martínez Maldonado I (2009). Ventajas del método de quimioluminiscencia frente al de radioinmunoanálisis (RIA). Vis ciento; 1 (2):60-68.
16. Fairbanks VF. Hemoglobinopathies and Talassemias. Edit B.C. Decker. USA 1980.
17. Fleming RE, Bacon BR. Orchestration of iron homeostasis. N Eng J Med 2005; 352: 1741-4.
18. Fleming DJ, Jacques PF, Tucker KL, Massaro JM, D’Agostino RB, Wilson WF, et al. Iron status of the free-living, elderly Framingham Heart Study cohort: an iron-

86
replete population with a high prevalence of elevated iron stores. Am J Clin Nutr 2001; 73: 638-46.
19. Goodnough LT, Skikne B, Brugnara C. Erythropoietin, iron, and erythropoiesis. Blood 2000; 96(3): 823-33.
20. Hernández R. 1996. Metodología de la investigación. Editorial Mc.Graw-Hill México. p. 35.
21. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 2004; 117: 285-97.
22. Jensen P-D. Evaluation of iron overload. Br J Haematol 2004; 124: 697-711. 23. Jeng MR, Adams-Graves P, Howard TA, Whorton MR, Li CS, Ware RE (2003).
Identification of hemochromatosis gene polymorphisms in chronically transfused patients with sickle cell disease. Am J Hematol; 74 (4): 243-248.
24. Kearney SM, Nemeth E, Neufeld EJ, Thapa D, Ganz T, Weistein DA, Cunningham MJ. Urinary hepcidin in congenital chronic anemias Pediat Blood Cancer 48: 57_63, 2007
25. Kwiatkowski JJ, Cohen AR. Iron chelation therapy in sickle-cell disease and other transfusion-dependent anemias. Hematol Oncol Clin N Am 2004;18:1355-1377)
26. Ladero J M.(2005) Hemocromatosis hereditaria de tipo I. Clínica,diagnóstico y tratamiento. Revista de la ACAD; I (3): 137-160.
27. Levi S, Corsi B, Bosisio M, Invernizzi R, Volz A, Sanford D, et al. A human mitochondrial ferritin encoged by an intronless gene. J Biol Chem 2001; 276: 24437-40.
28. Lieu PT, Heiskala M, Peterson PA, Yang Y. The roles of iron in health and disease. Mol Aspects Med 2001; 22: 1-87.).
29. Malcorra JJ. 2001. Hemoglobinopatías y Talasemias. Bscp can ped; 25 (2):265-277
30. Malcorra J. 2001 Hemoglobinopatías y Talasemias, BSCP Can Ped; 25 (2): 2265.267.
31. Milman N, Byg KE, Ovesen L, Kirchhoff M, Jurgensen KS. Iron status in Danish men 1984-1994: a cohort comparison of changes in iron stores and the prevalence of iron deficiency and iron overload. Eur J Haematol 2002; 68: 332-40.
32. Milman N, Byg KE, Ovesen L, Kirchhoff M, Jurgensen KS. Iron status in Danish women 1984-1994: a cohort comparison of changes in iron stores and the prevalence of iron deficiency and iron overload. Eur J Haematol 2003; 71:51-61.
33. Moirand R, Mortaji AM, Loreal O, Paillard F, Brissot P, Deugnier Y. A new syndrome of liver iron overload with normal transferrin saturation. Lancet 1997; 349: 95-7)
34. Muller H, Ziegler B, Schweizer B (1993). UV-VIS spectrometric methods for qualitative and and qualitative analysis of nucleic acids. Int Spectr Lab; 4: 4 – 11.
35. Napier I, Ponka P, Richardson DP. Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood 2005; 105: 1867-74.
36. Parkkila S, Parkkila A-K, Waheed A, Britton RS, Zhou XY, Fleming RE. 2000. Cell surface expression of HFE protein in epithelial cells, macrophages and monocytes. Haematologica; 85: 340-5
37. Papanikolaou G, Tzilianos M, Christakis JI, Bogdanos D, Simirika K, et al. Hepcidin in iron overload disorders Blood 105, 4103_4105, 2005
38. Pérez Surribas D, Viedma JA. Proteínas del metabolismo del hierro. Aplicaciones clínicas. Química Clínica 2006; 25 (1) 24-29

87
39. Pineda-Del Villar L, Borjas, M (1986). La hemoglobina S en la isla de Toas. Un problema genético de salud pública? Invest Clin; 27:5-14.
40. Pietrangelo A. Hereditary hemochromatosis – A new look at an old disease. N Eng J Med 2004; 350: 2383-97.
41. Powell LW, Jazwinska E, Halliday JW. Primary iron overload. In: Brock JH, Halliday JW, Pippard MJ, Powell LW (eds). Iron metabolism in health and disease. Philadelphia: WB Saunders, 1994: 227-70
42. Powell LW, George DK, McDonnell SM, Kowdley KV. Diagnosis of hemochromatosis. Ann Intern Med 1998; 129: 925-31)
43. Powell LW, Yapp TR. Hemochromatosis. Clin Liver Dis. 2000; 4:211-8. Adams PC. Hemochromatosis. Clin Liver Dis. 2004;8:735-53.
44. Ramsay WNM. The determination of the total iron-binding capacity of serum. Clin Chim Acta 1957; 2: 25-30.
45. Remacha AF, Carrasco M, Sardá MP, Barceló MJ, Blesa I, Baiget M. Screening for iron overload and HFE mutations in a university hospital. Haematologica 2000; 85: 873-4
46. Roa S, Martín J, Rodríguez R, García B, Sánchez A, Gónzalez R (2001). Estudio del gen HFE en una familia española con hemocromatosis hereditaria. Med. Clin (Barc.) ; 116: 100- 103).
47. Rodak F Bernadette (2004). Hematología: fundamentos y aplicaciones clínicas. 2da edición. Edit. Médica Panamericana, Buenos Aires. 217-218.
48. Roy CN, Andrews NC. Recent advances in disorders of iron metabolism: mutations, mechanisms and modifiers. Hum Mol Genet 2001; 10: 2182-6.
49. Salazar-Lugo R (2004). La hemoglobina S en la población Venezolana. Invest Clín; 45 (2): 175-183.
50. Sherwood RA, Pippard MJ, Peters TJ. Iron homeostasis and the assessment of iron status. Ann Clin Biochem 1998; 35: 693-708.
51. Tamayo y Tamayo Mario (2004). Proceso de la Investigación científica: Incluye evaluación y administración de proyectos de investigación. 4ta edición. México, D.F. Editorial Limusa-Noriega Editores: 47-49, 110-112,177-178.
52. Taher A, Sheikh-Taha M, Koussa S, Inati A, Neeman R, Mourad F.Comparison between deferoxamine and deferiprone (L1) in iron-loaded thalassemia patients. Eur J Haematol. 2001 Jul;67(1):30-4)
53. Tilg H, Ulmer H, Kaser A, Weiss G. Role of IL-10 for induction of anemia during inflammation. J Immunol 2002; 169: 2204-9.
54. Torres-Guerra E, Torres-Guerra T, Valbuena G, Arteaga Vizcaíno M, Soto L (1993). Frecuencia de anemia falciforme en la población de "Cuatro Bocas" parroquia Ricaurte, Municipio Mora, estado Zulia, Venezuela. Invest Clin; 34 (2):99-105.
55. Turgeon Mary Louise (2006). Hematología clínica. Teoría y procedimiento. Bogotá, D.C.Editorial Manual Moderno (Colombia),Ltda: 165 – 199.
56. U.S. Department of Health and Human Services. Division of Blood Disorders and Resources. The Management of Sickle Cell Disease. NIH publication No. 04-2117. Reprinted, June 2004
57. Wayne W Daniel. 1991. Bioestadística. Base para el análisis de las ciencias de la salud. 3era edición. Traductor: Manuel Guzmán Ortiz. México, D.F. Editorial. Noriega-Limusa: 503-507.
58. Weatherall DJ and JB Clegg. 1981. Thalassaemia Syndromes. 3ª edition. Blackwell.

88
59. Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med 2005; 352: 1011-23.
60. Worwood M. The laboratory assessment of iron status - an update. Clin Chim Acta 1997; 259: 3-23.

89
ANEXO

90 CONCENTRACION DE HIERRO ORGÁNICO EN ANEMIAS HEREDITARIAS COMO
PREVENCION DE HEMOSIDEROSIS
.HOJA DE RECOLECCION DE DATOS
Nombre y Apellido: _________________________________________
Fecha de Nac: _____________________ Edad: ____________ Sexo:__________
Dirección: __________________________________________________________
Estado: ___________________________ Municipio: ________________________
Diagnostico:________________________________________________
Ha recibido transfusión de concentrado globular: SI: ___ NO: ___
Especifique cantidad y el tiempo en que las ha recibido: ____________________
Ha recibido tratamiento con agentes quelantes de hierro: SI: ___ NO: ___
Niveles: hierro sérico: ________, SAT%:_________, TIBC: ______________
Ferritina sérica:__________
Laboratorio: Hb_____, Hcto______, Cta B_________, Plaq_____________
LDH_________, Fosfatasa Alcalina____________
HIV___________, VDRL______________
TGO: _____________ TGP: _______________
Examen.Fisico:__________________________________________________________


















