
Diabetes Insípida – Diagnóstico y Manejo · terminal de NPII (9 residuos aminoacídicos). El...
26
Minireview del Hormone Research in Paediatrics (con permiso) MINIREVISIÓN PUBLICADA EN HORMONE RESEARCH IN PAEDIATRICS TRADUCIDA DEL IDIOMA INGLÉS AL ESPAÑOL Este artículo es una traducción del original en Inglés, publicado en Horm Res Paediatr 2012;77:69-84. Para leer la versión en Inglés ir a www.karger.com/hrp. La publicación de esta traducción ha sido autorizada por S KARGER AG, Basel. Traducida por M. A. Rivarola y A. Belgorosky. Mini Revisión Horm Res Paediatr 2012;77:69–84 Diabetes Insípida – Diagnóstico y Manejo Natascia Di Iorgi a, Flavia Napoli a, Anna Elsa Maria Allegri a, Irene Olivieri a, Enrica Bertelli a, Annalisa Gallizia a, Andrea Rossi b, Mohamad Maghnie a, Departamentos de a Pediatría y b Pediatría Neuroradiológica, IRCCS G. Gaslini, Universidad de Génova, Génova, Italia Resumen La diabetes insípida central (DIC) es el resultado final de un número de condiciones que afectan al sistema hipotalámo-neurohipofisario. Las causas conocidas incluyen los germinomas/craneofarengiomas, histiocitosis de células de Langerhans (HCL), enfermedades inflamatorias locales, autoinmunes o vasculares, trauma quirúrgico o por accidente, sarcoidosis, metástasis y malformaciones craneales o de la línea media del cerebro. En casos raros, la causa subyacente puede ser defectos genéticos en la síntesis de vasopresina que se heredan en forma autosómica dominante, autosómica recesiva o ligada al X. El diagnóstico de la condición subyacente es un desafío y crea preocupación en los pacientes y sus padres porque demanda un seguimiento prolongado. El diagnóstico etiológico apropiado requiere una serie de pasos que comienzan con observaciones clínicas y luego progresan a herramientas más sofisticadas. Específicamente, la identificación por RMN de una hiperintensidad hipofisaria en la parte posterior de la silla turca, considerada ahora un marcador claro de integridad funcional neurohipofisaria, junto con un análisis cuidadoso de la forma y tamaño del tallo hipofisario han contribuido en forma notoria al diagnóstico y comprensión de algunas formas DIC. La MRI STIR es una tecnología prometedora para la identificación temprana de la CDI dependiente de HCL. Definición/Clasificación La diabetes insípida es una enfermedad en la cual se excretan grandes volúmenes de orina diluida (poliuria) debido a deficiencia de vasopresina (AVP) [diabetes insípida central (DIC)], resistencia a AVP [diabetes insípida nefrogénica (DIN)], o
-
Upload
nguyendung -
Category
Documents
-
view
215 -
download
0
Transcript of Diabetes Insípida – Diagnóstico y Manejo · terminal de NPII (9 residuos aminoacídicos). El...

Minireview del Hormone Research in Paediatrics (con permiso)
MINIREVISIÓN PUBLICADA EN HORMONE RESEARCH IN PAEDIATRICS TRADUCIDA DEL IDIOMA INGLÉS AL ESPAÑOL
Este artículo es una traducción del original en Inglés, publicado en Horm Res Paediatr 2012;77:69-84. Para leer la versión en Inglés ir a www.karger.com/hrp. La publicación de esta traducción ha sido autorizada por S KARGER AG, Basel. Traducida por M. A. Rivarola y A. Belgorosky. Mini Revisión
Horm Res Paediatr 2012;77:69–84 Diabetes Insípida – Diagnóstico y Manejo Natascia Di Iorgi a, Flavia Napoli a, Anna Elsa Maria Allegri a, Irene Olivieri a, Enrica Bertelli a, Annalisa Gallizia a, Andrea Rossi b, Mohamad Maghnie a, Departamentos de a Pediatría y b Pediatría Neuroradiológica, IRCCS G. Gaslini, Universidad de Génova, Génova, Italia Resumen La diabetes insípida central (DIC) es el resultado final de un número de condiciones que afectan al sistema hipotalámo-neurohipofisario. Las causas conocidas incluyen los germinomas/craneofarengiomas, histiocitosis de células de Langerhans (HCL), enfermedades inflamatorias locales, autoinmunes o vasculares, trauma quirúrgico o por accidente, sarcoidosis, metástasis y malformaciones craneales o de la línea media del cerebro. En casos raros, la causa subyacente puede ser defectos genéticos en la síntesis de vasopresina que se heredan en forma autosómica dominante, autosómica recesiva o ligada al X. El diagnóstico de la condición subyacente es un desafío y crea preocupación en los pacientes y sus padres porque demanda un seguimiento prolongado. El diagnóstico etiológico apropiado requiere una serie de pasos que comienzan con observaciones clínicas y luego progresan a herramientas más sofisticadas. Específicamente, la identificación por RMN de una hiperintensidad hipofisaria en la parte posterior de la silla turca, considerada ahora un marcador claro de integridad funcional neurohipofisaria, junto con un análisis cuidadoso de la forma y tamaño del tallo hipofisario han contribuido en forma notoria al diagnóstico y comprensión de algunas formas DIC. La MRI STIR es una tecnología prometedora para la identificación temprana de la CDI dependiente de HCL. Definición/Clasificación La diabetes insípida es una enfermedad en la cual se excretan grandes volúmenes de orina diluida (poliuria) debido a deficiencia de vasopresina (AVP) [diabetes insípida central (DIC)], resistencia a AVP [diabetes insípida nefrogénica (DIN)], o

ingesta de agua excesiva (polidipsia primaria). La poliuria se caracteriza por un volumen urinario en exceso de 2 L/m2/24 h, o aproximadamente 150 ml/Kg/24 h al nacimiento, 100-110 ml/Kg/24 h hasta la edad de 2 años y 40-50 ml/Kg/24 h en niños mayores y adultos. Etiología En muchos pacientes, la DIC está causada por la destrucción o degeneración de neuronas que se originan en los núcleos supraóptico y paraventricular. Las causas conocidas de estas lesiones incluyen enfermedades locales autoinmunes o inflamatorias, enfermedades vasculares, histiocitosis de células de Langerhans (HCL), sarcoidosis, germinoma/craneofaringeoma, trauma resultante de cirugía o accidente y malformaciones de línea media cerebrales o craneales [1]. En casos raros, las causas subyacentes son defectos genéticos en la síntesis de AVP, heredados en forma autosómica dominante, autosómica recesiva, o recesiva ligada al X. La DIN recesiva ligada al X es secundaria a mutaciones del receptor de AVP 2 (AVPR2), que resulta en pérdida de función o desregulación del AVPR2 renal. Las anormalidades del gen del canal de agua acuaporina 2 (AQP2), localizado en el cromosoma 12 a nivel q13, son responsables de formas autosómicas familiares, recesivas y dominantes, de DIN. Epidemiología La diabetes insípida es una enfermedad rara con una prevalencia reportada de 1: 25,000 [2]. Menos de 10% de la diabetes insípida puede atribuirse a formas hereditarias [3]. En particular, la DIN ligada al X (OMIM 3048000) representa el 90% de los casos de DIN congénita y se encuentra con una frecuencia de 4-8 por millón de nacimientos de varones vivos, mientras que la DIN autosómica (OMIM 125800) constituye aproximadamente el 10% de los casos restantes [4]. No hay diferencias de género en las otras formas. Aunque ha sido reportado que la prevalencia del síndrome de Wolfram es 1-9 en 1,000,000 (www.orpha.net) la frecuencia de la DIC aún se desconoce. Patogénesis y Patología Anatomía La hipófisis posterior está formada por neuronas magno-celulares que producen AVP y/u oxitocina. Los cuerpos celulares de las neuronas magno-celulares están localizados en los núcleos paraventricular y supraóptico y sus axones se proyectan a la neurohipófisis desde donde las hormonas se segregan al torrente sanguíneo. Estos axones almacenan suficientes cantidades de AVP como para sostener una liberación basal durante 30-50 días, o permitir una antidiuresis máxima por 5-10 días [5]. Mientras que la circulación sanguínea de la hipófisis anterior se realiza vía el sistema porta hipotalámico-hipofisario desde las arterias suprahipofisarias, la vascularización de la hipófisis posterior es directa desde las arterias hipofisarias inferiores. Biosíntesis de AVP El gen AVP-neurofisina II (AVP-NPII) está localizado en el brazo corto del cromosoma 20 (20p13). Cubre 2,5 kb y comprende 3 exones. El exón 1 codifica el péptido señal de 19 residuos aminoacídicos, el nonapéptido AVP y la región N-

terminal de NPII (9 residuos aminoacídicos). El exón 2 codifica para la altamente conservada región central del péptido NPII (67 residuos de aminoácidos). El exón 3 codifica para la región C-terminal del NPII (17 residuos de aminoácidos) y un glucopéptido de 39 aminoácidos llamado copeptina {6}. El producto del gen AVP-NPII, la preprehormona se co-traduce y dirige al retículo endoplásmico (RE), donde el péptido señal se libera por acción de una peptidasa y el copéptido se glicosila en su núcleo. El AVP y el copéptido se asocian luego del clivaje por la señal-péptidasa y entonces forman el tetrámero que aumenta la afinidad de unión de AVP con NPII. Luego de la formación de uniones 7 disulfuro dentro de NPII y una dentro de AVP y luego de glicosilación del copéptido, el proprecursor se empaqueta en los gránulos neurosecretorios y se cliva a los productos peptídicos durante el transporte axonal hacia la hipófisis posterior [6]. La neurofisina sirve para estabilizar a la hormona durante el trasporte y almacenamiento, mientras que datos recientes sugieren que la copeptina podría jugar un rol importante en la formación estructural correcta del precursor de AVP como un prerrequisito para su maduración proteolítica eficiente {7}. La AVP y su proteína trasportadora NPII son liberadas de la hipófisis posterior cuando el axón es despolarizado por los estímulos de osmoreceptores o baroreceptores (Fig. 1). Fisiología de la homeostasis del agua. El mantenimiento del balance de agua en los humanos sanos se logra principalmente por tres determinantes interrelacionados: la sed, la AVP y la función renal. Recientemente, se ha aislado la apelina de extractos de estómago bovino, un péptido bioactivo (como en el caso de la ghrelina, es otra asociación estómago-hipotálamo). Se expresa en los núcleos supraóptico y paraventricular y ejerce su acción en receptores específicos localizados en neuronas vasopresinérgicas. La apelina actúa como un neuropéptido diurético potente que contrarresta las acciones de AVP a través de la inhibición de su actividad neuronal y liberación. La coexistencia de apelina y AVP en las neuronas magnocelulares, junto con sus efectos biológicos contrapuestos es probable que jueguen un rol clave en el mantenimiento de los fluidos corporales [8]. La AVP actúa sobre su órgano blanco mayor, el riñón, donde aumenta la osmolaridad de la orina. Se une a los receptores V2 en la membrana basolateral del tubo colector renal y activa el sistema Gs-adenil ciclasa, aumentando los niveles intracelulares de 3´,5´-adenosina monofosfato cíclico (cAMP). Este último activa a la proteína kinasa A, la que a su vez fosforila al canal de agua AQP2 preformado y localizado en vesículas intracelulares [9]. La fosforilación promueve el tráfico a la membrana apical, seguido de inserción exocítica de las vesículas AQP2 en la membrana celular. La inserción de AQP2 hace que el tubo colector se vuelva permeable al agua, permitiendo el libre movimiento de agua desde el lumen del nefrón al interior de las células del tubo colector y generando un gradiente osmótico, es decir concentrando la orina. La síntesis de los canales AQP2 y su movimiento es regulada por la estimulación de AVP. La acuaporina 3 y la acuaporina 4 responsables del pasaje subsiguiente de agua desde el interior de las células al intersticio, están presentes en forma constitutiva en la membrana basolateral [10].

Figura 1. Representación esquemática de la biosíntesis de AVP. CO = Copeptina; NP = neurofisina Patogenia Se produce un aumento de la poliuria cuando se dañan más del 80% de las neuronas que segregan AVP. La destrucción extensa puede ser causada por varios procesos patológicos, incluyendo causas genéticas. Por estudios de autopsia se conoce que luego de la sección del tallo hipofisario (TH) por traumatismo se produce una gran pérdida de las células neurosecretorias en los núcleos hipotalámicos. Esto se produce dentro de las 4-6 semanas, con mayor daño para las lesiones a nivel del infubdíbulo o por encima de éste [11]. La autopsia de pacientes con una forma familiar de diabetes insípida muestra una pérdida selectiva de neuronas del núcleo paraventricular asociada a una gliosis moderada y una preservación relativa de células neurosecretoras pequeñas [12], sugiriendo que la alteración es secundaria a la degeneración de estas neuronas hipotalámicas; Formas genéticas de DIC Hasta el momento se han identificado más de 55 mutaciones diferentes que resultan en una pro-hormona deficiente y una deficiencia de AVP en la DIC familiar neurohipofisaria [6, 13]. Casi todas tienen un característica hereditaria autosómica dominante, pero se han reportado seis pacientes con una mutación homocigota sin sentido en la región que codifica para el dominio AVP que muestran una característica de herencia autosómica dominante [14, 15]. Si bien posee algunas similitudes con la forma dominante, los síntomas en estos casos parecen deberse a una actividad biológica reducida del péptido AVP mutado. Esta hipótesis se apoya en los niveles elevados de hormona circulante, la ausencia de hormona AVP normal en el estado homocigota, y en la ausencia de anormalidades clínicas

o subclínicas en portadores heterocigotas. No se encontró mutación en la región codificante, la región intrónica o la región de 1,5-kb corriente arriba del sitio de iniciación de la transcripción del gen AVP-NPII en una familia china con un patrón de herencia autosómico dominante de DIC severa [16]. El análisis de linkage indicó que los gene(s) responsable(s) de la forma autosómica dominante en esta familia estaba(n) localizado(s) en un intervalo de 7 cM definido por dos marcadores cortos en tándem en el cromosoma 20. Esto sugiere la presencia de heterogeneidad del locus genético de la DIC autosómica dominante e implica una diversidad genética en las causas de la DIC. La herencia autosómica dominante de esta enfermedad puede ocurrir por varios mecanismos incluyendo actividad negativa dominante por interacción del precursor salvaje y mutado, acumulación de precursor mutado en el retículo endoplásmico (ER) que lleve a una respuesta de estrés proteico y autofagia y toxicidad celular por mecanismos aun no definidos completamente. El estudio del tráfico y procesamiento de la prohormona AVP mutada in vitro ha demostrado que la mutación elimina la salida y procesamiento de la prohormna AVP en el RE resultando en una morfología endoplásmica aberrante y posiblemente disfunción y muerte celular [6]. La presencia de autofagia citosólica sugiere muerte no apoptótica [17-19]; sin embargo, no se puede excluir la muerte celular programada [20]. Las mutaciones que involucran al péptido señal disminuyen su habilidad para iniciar el adecuado procesamiento de la prepro-AVPNPII [21]. Los precursores mutados también impiden el tráfico intracelular del precursor salvaje formando heterodímeros y de esta manera reduciendo la biodisponibilidad de la AVP activa por medio de un mecanismo “no tóxico”, esto es, un efecto dominante negativo [19, 22]. La demostración de dos vías de degradación (vía el lumen del ER y directamente en el citosol) involucrando tanto la prohormona salvaje como la mutada sugiere que el efecto citotóxico podría resultar de procesos que son cuantitativamente, pero no fundamentalmente diferente de aquellos que suceden en las células que expresan la proteína salvaje [21, 23]. Formas Adquiridas de CDI CDI Idiopática Aunque 20-50% de los casos son considerados “idiopáticos”, la identificación de anticuerpos contra las células secretorias de AVP (anti-cAVP) por una parte, y los avances recientes en las técnicas de imágenes [24] por otra, han arrojado nuevas luces sobre los aspectos patofisiológicos de la DIC, haciendo que la forma idiopática se vuelva una condición muy poco común. Varias observaciones clínicas sugieren que la inmunidad tiene un rol importante en la patogénesis de la DIC. La poliendocrinopatía autoinmune y la DIC se asocian con una imagen de engrosamiento del TH sugiriendo que comparten una etiología común [1, 25, 26]. Debido a que hay una relación temporal entre una infección viral (disparador) y el comienzo de DIC en aproximadamente un cuarto de los pacientes con DIC idiopática [1], ésta participación de la hipófisis anterior en la evolución de la DIC idiopática hace altamente sospechosa la existencia de una base neurohipofisaria autoinmune, lo que se ajusta muy bien con la demostración de infiltración linfocítica en el TH [27]. Esta hipótesis se ve reforzada por el hecho de que la glándula hipofisaria es susceptible de autoinmunidad mediada por

células T CD8, desencadenada por un modelo autoantígeno de especificidad celular [28], así como también por el desarrollo de hipofisitis autoinmune luego de la inmunización del ratón SJL/J con extractos de hipófisis de ratones [29]. Sin embargo, la identificación de anti-cAVP en sujetos que tienen DIC idiopática, HCL o germinoma indica que este hallazgo no puede ser considerado un marcador completamente confiable de DIC idiopática [24]. Por lo tanto, para asegurar un diagnóstico etiológica definitivo, se necesita un cuidadoso seguimiento clínico y con RMN, ya que la presencia de anti-cAVP podría enmascarar a un germinoma o a una HCL. El proceso subyacente de engrosamiento del TH en la DIC idiopática no se comprende completamente. El término “infundíbulo-hipofisitis linfocítica” fue propuesto para distinguir entre los niños y adolescentes con DIC, deficiencia hormonal hipofisaria, reducción del tamaño de la hipófisis anterior, engrosamiento del TH transitorio o persistente y pacientes adultos con hallazgos en el TH similares en la RMN, pero hipófisis anterior de tamaño y función normales [30]. La identificación por parte de Mirocha y co. [31] de dos mecanismos patogénicos potenciales diferentes, uno potencialmente dirigido a contra auto-antígenos (dominación de T helper) y otro no (post-infección) ambos induciendo hipofisitis primaria agrega otra pieza al rompecabezas de esta intrigante condición. Los pacientes con hipofisitis secundaria a autoinmunidad se beneficiarían con esteroides u otro tratamiento inmunosupresor, mientras que la inmunosupresión podría exacerbar condiciones que no son auto-inmunes. Vale la pena señalar que localizaciones extra-hipofisarias de la HCL, incluyendo el tórax o el hígado pueden aparecer luego del comienzo de la DIC [32]. Raramente, mutaciones de novo del gen AVP-NPII son responsables de algunas formas idiopáticas de DIC asociadas con tamaño normal del TH [33, 34]. DIC vascular La DIC puede ser causada por un daño vascular cerebral, pero la patofisiología de este mecanismo nunca ha sido comprendida con precisión. En un grupo de pacientes con DIC idiopática y función hipofisaria anterior normal, la RMN estándar mostró una glándula de tamaño normal con TH normal [35]. Más aún, estudios de RMN dinámica luego de la inyección de medio de contraste revelaron ausencia de refuerzo del lóbulo posterior de la hipófisis, mientras que hubo refuerzo normal en la hipófisis anterior. La falta de refuerzo del contraste del lóbulo de la hipófisis posterior sugiere un daño vascular selectivo de las arterias hipofisarias inferiores que podría ser la causa de la DIC. No se ha definido cual es el mecanismo que podría afectar el flujo sanguíneo de la hipófisis posterior, pero la posibilidad de que una alteración congénita del desarrollo del sistema vascular de la hipófisis posterior (sin evidencias de anormalidades macroscópicas de la glándula hipofisaria en la RMN o cambios secundarios del flujo vascular secundarios a procesos inflamatorios locales, por ejemplo, vasculitis), no puede ser descartada. Histiocitosis de Células de Langerhans La DIC es la manifestación de la HCL en el SNC, con una frecuencia de 10-50% [36, 37]. Un análisis multicéntrico retrospectivo de pacientes con HCL mostró que el riesgo de desarrollar DIC, luego del diagnóstico y terapia específica, era 16% a los 5 años de edad y 20% a los 15 años de edad, respectivamente, y que

correlacionaba fuertemente con la presencia de enfermedad multisistémica seguida de lesiones en el área craneofacial [36]. Algunos pacientes con DIC y endocrinopatías parecen estar en riesgo de enfermedad neurodegenerativa del SNC de larga evolución, aunque participación del cerebro y la pineal puede ser diagnosticada precozmente luego del inicio de la DIC. La deficiencia de hormona de crecimiento es el déficit adicional más frecuente, estando presente en el 42% de los casos de DIC e HCL. La incidencia acumulativa de deficiencia de hormona de crecimiento en 10 años entre pacientes con DIC del estudio francés sobre HCL en todo el país fue de 54% [38]. La identificación de anti-cAVP en pacientes con HCL y su tendencia a la desaparición espontánea [24, 25] sugiere que estos anticuerpos podrían ser un epifenómeno relacionado con la HCL. El engrosamiento del TH puede ser encontrado en aproximadamente 50-70% de los pacientes con HCL a la presentación o en el seguimiento [1, 39] y puede aún estar presente antes del comienzo de la DIC. El tamaño de la hipófisis anterior puede estar normal, reducido o, raramente, aumentado [1, 36, 40]. Se recomienda la búsqueda de lesiones extracraneales (dermatológicas, óseas, en radiografías de tórax, y en el examen de garganta, nariz y oído) sugestivas de HCL en pacientes con engrosamiento del TH ya que podría reducir la necesidad de la biopsia intracraneal. Tumores Germinomas Los tumores germinales intracraneales comprenden el 7,8% de los tumores pediátricos de cerebro [41]. Los hallazgos de RMN sugieren que los germinomas supraselares y neurohipofisarios derivan primariamente desde la hipófisis posterior al infundíbulo [42]. En un 78-100% de los casos se detecta engrosamiento parcial o completo del TH a la presentación y puede llegar a ser el único hallazgo en germinomas pequeños [42]; su presencia aumenta el riesgo de malignidad a alrededor de 15-17%, mientras que el riesgo disminuye a 3% en los pacientes con un TH de tamaño normal. La realización de RMN de cerebro con contraste seriada en pacientes afectados por DIC con TH engrosado (cada 3-6 meses durante los primeros 2 años) puede reducir el tiempo de diagnóstico del germinoma hasta en 1 año [1]. Sin embargo, el engrosamiento del TH ha sido reportado hasta 5 años después del comienzo de la DIC, precedida por infiltración linfocitaria del tejido como reacción de huésped a la presencia de germinoma, lo que puede enmascarar el diagnóstico [43]. Como excepción, el germinoma puede simular una HCL multisistémica, con compresión vertebral, infecciones del oído recurrentes, engrosamiento del TH, agrandamiento de la glándula pineal y marcadores de tumor de células germinales negativos en suero y líquido céfaloraquídeo, como se ha demostrado en una niña de 9 años de edad [44]. El rol de la determinación de gonadotrofina coriónica humana (hCG) y otros marcadores tumorales en el diagnóstico precoz del germinoma no está bien comprendido. Un resultado negativo de hCG en líquido cefaloraquídeo no descarta el diagnóstico de germinoma [1]. La presencia de anti-cAVP en la circulación en estos pacientes antes del tratamiento [24] puede también enmascarar el diagnóstico de germinoma y requerir confirmación adicional. La biopsia de de TH es mandatoria en presencia de un engrosamiento progresivo de la lesión hasta

más de 6,5-7 mm y/o agrandamiento de la hipófisis. La detención del crecimiento y la deficiencia hipofisaria múltiple son hallazgos tempranos y frecuentes (casi el 100% de los casos en el seguimiento), pero la deficiencia hormonal no es necesariamente predicción de germinoma. Craneofaringeoma y DIC post-cirugía. El craneofaringeoma es un tumor benigno que se origina en los nidos de células escamosas de la bolsa de Rathke primitiva. Constituye aproximadamente el 6-9% de los tumores intracraneanos en los niños y es el neoplasma supraselar más frecuente en la población pediátrica, alrededor del 54% de los casos [41]. Las presentaciones clásicas incluyen alteraciones visuales, debida a la compresión del quiasma y atrofia óptica bilateral; los síntomas sistémicos relacionados con el aumento de la presión intracraneana se presentan en el 60-75% de los casos [41]. En varias series pediátricas grandes, se reportan signos y síntomas de disfunción de la hipófisis anterior en alrededor del 20-70% de los casos. La DIC y la deficiencia hipofisaria múltiple son complicaciones comunes en el craneofaringeoma de la niñez. La frecuencia de la DIC pre-quirúrgica varía alrededor de 16-55%, mientras que la frecuencia post-quirúrgica y permanente se onserva en hasta el 80% de los casos; La DIC transitoria se ha reportado en el 13% de casos afectados [45]. Luego de la sección del TH, un resultado predecible es la deficiencia de la función de la hipófisis posterior, caracterizada por la respuesta trifásica del volumen urinario. La fase inicial de DIC (1-4 días) es seguida por una segunda fase de oliguria que podría reflejar la degeneración y muerte de las neuronas neurosecretorias con la liberación de AVP almacenada a la circulación (4-7 días), y una tercera y última fase de DIC permanente. El diagnóstico de la DIC se hace frecuentemente unas pocas horas después de la cirugía, aunque las anormalidades de la secreción de AVP y del balance de los líquidos comienza frecuentemente en el período intra-operatorio [45]. Un estudio reciente demostró que la respuesta trifásica luego de la cirugía primaria por craneofaringeoma en niños se puede predecir en las cirugías de larga duración [46]. Diagnóstico de Diabetes Insípida Manifestaciones Clínicas. Síntomas y Signos El examen clínico puede proveer claves importantes al posible diagnóstico subyacente. La edad en que aparecen los síntomas junto con las características de la ingestión de líquidos puede influir sobre la investigación de diabetes insípida subsiguiente. Los síntomas primarios son poliuria y polidipsia, y los niños pequeños pueden tener deshidratación severa, vómitos, constipación, fiebre, irritabilidad, trastornos del sueño, falta de progreso de peso y retardo de crecimiento. La nocturia de los niños se presenta frecuentemente como enuresis. Una deshidratación severa de comienzo temprano en varones es altamente sugestiva de DIC; se ha reportado algún retardo mental, probablemente causado por una deshidratación no reconocida antes de que el diagnóstico haya sido establecido. En una cohorte grande de pacientes con DIC de diferentes etiologías [1], 40% de los pacientes tenían síntomas aparte de poliuria y polidipsia a la presentación; mientras que la cefalea no era discriminatoria, los defectos visuales se asociaron a tumor intracraneal. El retardo de crecimiento no fue

significativamente más común en pacientes con tumores del SNC, en contraste con reportes previos que indicaban que estos retardos sugerían fuertemente que un tumor intracraneal era la causa de la DIC. Además, los pacientes que no tenían tumor intracraneal, eran significativamente menores de 5 años de edad [1]. En la DIC autosómica dominante, el comienzo de la enfermedad típicamente varía entre 1 y 6 años de edad, pero se han reportado varios casos de comienzo temprano o tardío [47]. Habitualmente, los síntomas empeoran con la edad en pacientes con comienzo temprano de poliuria y polidispsia especialmente antes de los 10 años de edad, pero es también posible que la DIC completa se exprese desde el período neonatal [41]. La gran variabilidad en la edad de comienzo y en la severidad de la deficiencia de AVP entre los pacientes con la misma mutación puede atribuirse a diferencias individuales entre estos pacientes, tales como la cantidad de producción del precursor mutado, la intensidad de la estimulación neurohipofisaria, la susceptibilidad a los efectos tóxicos del precursor mutado, la capacidad de degradar precursores mutados y las variaciones en la capacidad de reserva secretoria o en el desarrollo de la glándula misma. En el síndrome de Wolfram, se ha reportado que la diabetes mellitus es usualmente el primer síntoma que se presenta a una edad media de 6 años, seguida por el comienzo de atrofia óptica a una edad media de 11 años [48]. La correlación fenotipo-genotipo en una serie de 9 familias con síndrome de Wolfram muestran una edad promedio de comienzo de diabetes mellitus de 8,4 años, en concordancia con otros estudios [49, 50]. El desarrollo de poliuria y/o enuresis puede indicar diabetes insípida, y el momento del comienzo varía considerablemente, pero generalmente no aparece hasta la segunda o tercera década de la vida [39, 46]; la DIC puede ser inicialmente parcial. La frecuencia de DIC varía entre publicaciones entre 48 y 78% [51]. Medición de la Osmolaridad Mientras que la tonicidad y osmolaridad del sodio y otros electrolitos son idénticas, la urea y la glucosa muestran gran variación entre la presión osmótica determinada por disminución del punto de congelamiento y la osmolaridad efectiva in vivo. La precisión de la medición de la osmolaridad del plasma en los laboratorios hospitalarios de rutina, evaluada por disminución del punto de congelamiento, no es habitualmente alta como para satisfacer los criterios de calidad requeridos (coeficiente de variación de 1% a 290 mosm/Kg H2O), especialmente cuando la osmolaridad es determinada en suero o plasma congelado. Pero la osmolaridad extracelular y la plasmática pueden razonablemente considerarse que se corresponden con las sales de sodio. Por lo tanto, la osmolaridad del plasma en (mosm/Kg H2O) puede ser groseramente calculada como el doble de la concentración de sodio (en mEq/l). Sin embargo, la contribución de otros dos solutos, glucosa y urea, debería ser incluida para estimar con más precisión la osmolaridad del plasma: 2 [Na+] + [glucosa en sangre] + [urea]. El peso molecular de la glucosa es 180 y el de nitrógenos en la urea de 28. El contenido en plasma de ambos se expresa habitualmente como mg/dl (en lugar de mg/l), de tal manera que los pesos moleculares deben dividirse por 10. Por lo tanto, la glucosa se puede estimar por el contenido de glucosa del plasma (en mg/dl)/18, y la urea por el nitrógeno de urea en sangre (BUS en inglés; en mg/dl)/2,8. Es obvio que los laboratorios que usan unidades SI no deben hacer los

cálculos mencionados anteriormente. Finalmente, una buena manera de estimar la osmolaridad del plasma dentro del 1-3% (por ejemplo 9 mosm/Kg H2O) de lo que es determinado directamente por osmometría [11], puede obtenerse con la fórmula siguiente: P= 2[Na+]+(glucose/18)+(BUN/2,8). Test de Deprivación y desafío con Desmopressin Es esencial completar una colección de orina de 24 horas y medir su volumen antes de confirmar la presencia de poliuria. Para un diagnóstico correcto se requiere una serie de investigaciones básicas incluyendo electrolitos en plasma, la osmolaridad en plasma al azar, y la osmolaridad urinaria, así como una evaluación de la función renal. En ausencia de un diagnóstico inmediato, se debe estudiar con mayor detalle la ingesta y eliminación de líquidos. Se debe establecer la capacidad del SNC de producir AVP y la del riñón de responder a ésta utilizando el test formal de privación de agua y la respuesta a la desmopressina (DDAVP) [52]. Generalmente es suficiente con un test 7 horas (o menos) de privación de agua para el diagnóstico, excepto en casos de polidipsia primaria donde a veces se requiere un tiempo mayor. El test debe ser interrumpido si la pérdida de peso excede 5% del peso inicial y/o el N+ plasmático supera el valor de 143 mEq/l y/o la osmolaridad del plasma supera el valor de 295 mosm/kg H2O, y/o la osmolaridad en orina aumenta a lo normal (tabla 1). Es importante tener los resultados rápidamente, por lo que se necesita una colaboración estrecha con el laboratorio. El diagnóstico de DIC se basa en la demostración de hiperosmolaridad plasmática (>300 mosm/l) asociada a hipoosmolaridad urinaria (<300 mosm/l o una relación osmolaridad urinaria/plasmática <1), y poliuria (volumen urinario >4-5 ml/kg/h durante 2 horas (consecutivas) luego de la cirugía. La deficiencia de ACTH puede enmascarar los signos de DIC parcial, y la poliuria sólo manifestarse luego de la terapia de reemplazo con corticoides [45]. La administración de DDAVP ayuda a hacer el diagnóstico diferencial entre DIC y DIN. Recientemente, la copeptina y la AQP2 también han sido usadas en el diagnóstico diferencial de CDI versus DIN [53, 54]. La aquaporina se sintetiza en el riñón y es excretada en la orina en respuesta a la AVP. Los paciente con DIC no muestran aumento de AQP2 con la deshidratación, pero su excreción aumenta en respuesta a DDAVP, sugiriendo que la expresión de AQP2 persiste en pacientes con DIC. Por lo tanto, el principal valor de la AQP2 en el diagnóstico diferencial de la diabetes insípida sería indicar un diagnóstico de DIN cuando no hay ningún incremento en la excreción de AQP2 luego de la administración de DDAVP. Tabla 1. Interpretación del test de deprivación y del test de DDAVP Osmolaridad urinaria, mosm/kg
Osmolaridad urinaria, mosm/kg
DIAGNOSTICO
Post deprivación de agua Post DDAVP ˂300 ˃750 DIC ˂300 ˂300 DIN ˃750 --- PP 300-750 ˂750 DIC parcial ? DIN parcial ?

Polidipsia primaria ?
Estudios de Imágenes Una vez que se ha establecido el diagnóstico de DIC, son obligatorios otros estudios para buscar la causa, incluyendo marcadores tumorales, búsqueda de lesiones esqueléticas (en la HCL, la lesión del cráneo está presente hasta en un 85% de los casos), y especialmente neuroimágenes del cerebro [13]. En la RMN la hipófisis anterior puede verse como una señal hiperintensa en la imagen sagital con peso de T1 en condiciones basales (Fig. 2). La falta de señal hiperintensa (aunque no específica) es una marca clásica de trastorno hipótalmico-hipófisoposterior y puede significar presencia temprana de tumor local oculto (Fig. 3). En la DIC dominante, la identificación de la hiperintensidad de la hipótesis posterior no indica necesariamente integridad funcional del eje hipotalamo-neurohipofisis [1, 13, 39, 55]. Cuando está presente inicialmente, la señal desaparece en forma regular durante el seguimiento. El engrosamiento del TH o infundíbulo, definido por exceder 3-mm comparado con la parte distal, aunque no es específico (a veces la parte proximal del tallo tiene menos de 3 mm de espesor comparando con la parte distal) se observa en aproximadamente un tercio de los niños con DIC (Fig. 4). El tamaño del TH al inicio es variable y puede cambiar con el tiempo (Fig. 5). En dos grandes series pediátricas de pacientes con DIC idiopática, se encontró engrosamiento del TH en aproximadamente 50-60% de los sujetos [1, 40]. La evolución espontánea del TH engrosado fue similar en ambos reportes, de sin cambios (30%), a reducción (30-50%), o aumento mayor (10-20%) del tamaño del tallo. Entre los pacientes con DIC idiopática y un TH engrosado, un 90-94% de ellos desarrollaron deficiencias de las hormonas de la hipófisis anterior, de las que la deficiencia de la hormona de crecimiento representó el 60% de los casos. La deficiencia de hormonas hipofisarias múltiple estuvo presente en 30-50% de los pacientes con engrosamiento hipofisario mientras que solamente el 10% de 19 pacientes con TH normal tuvieron déficits hormonales adicionales [1]. Es importante el examen del cerebro para descartar localizaciones en el SNC, así como si existe otitis media recurrente, o disnea para descartar enfermedad multiorgánica (Fig. 6). Se necesita un seguimiento clínico, radiológico y de estudios endócrinos. En particular se recomienda seguimiento con RMN en todos los pacientes con engrosamiento del TH (cada 3-6 meses) y biopsia del TH si la RMN revela agrandamiento de la lesión del TH (>6,5 mm) o de la glándula hipofisaria posterior (el tamaño de la hipófisis anterior es dependiente de la edad), o afectación del tercer ventrículo (Fig. 7, tabla 2) [1, 13, 27, 40-42, 56, 57]. La RMN dinámica puede ayudar a identificar casos de DIC y tamaño normal del TH asociada a irrigación sanguínea anormal de la hipófisis posterior [33, 58]. En las figuras 8 y 9 se muestran esquemas diagnósticos para la DIC.
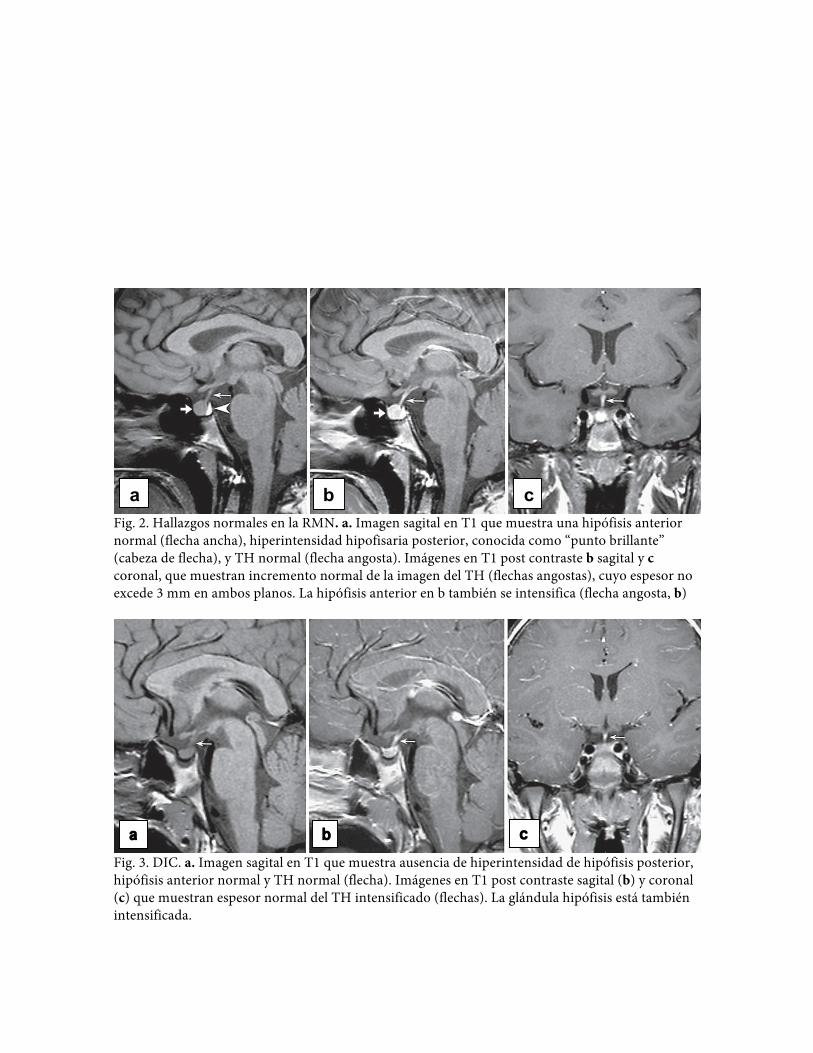
Fig. 2. Hallazgos normales en la RMN. a. Imagen sagital en T1 que muestra una hipófisis anterior normal (flecha ancha), hiperintensidad hipofisaria posterior, conocida como “punto brillante” (cabeza de flecha), y TH normal (flecha angosta). Imágenes en T1 post contraste b sagital y c coronal, que muestran incremento normal de la imagen del TH (flechas angostas), cuyo espesor no excede 3 mm en ambos planos. La hipófisis anterior en b también se intensifica (flecha angosta, b)
Fig. 3. DIC. a. Imagen sagital en T1 que muestra ausencia de hiperintensidad de hipófisis posterior, hipófisis anterior normal y TH normal (flecha). Imágenes en T1 post contraste sagital (b) y coronal (c) que muestran espesor normal del TH intensificado (flechas). La glándula hipófisis está también intensificada.
a b c
a b c

a b
c d
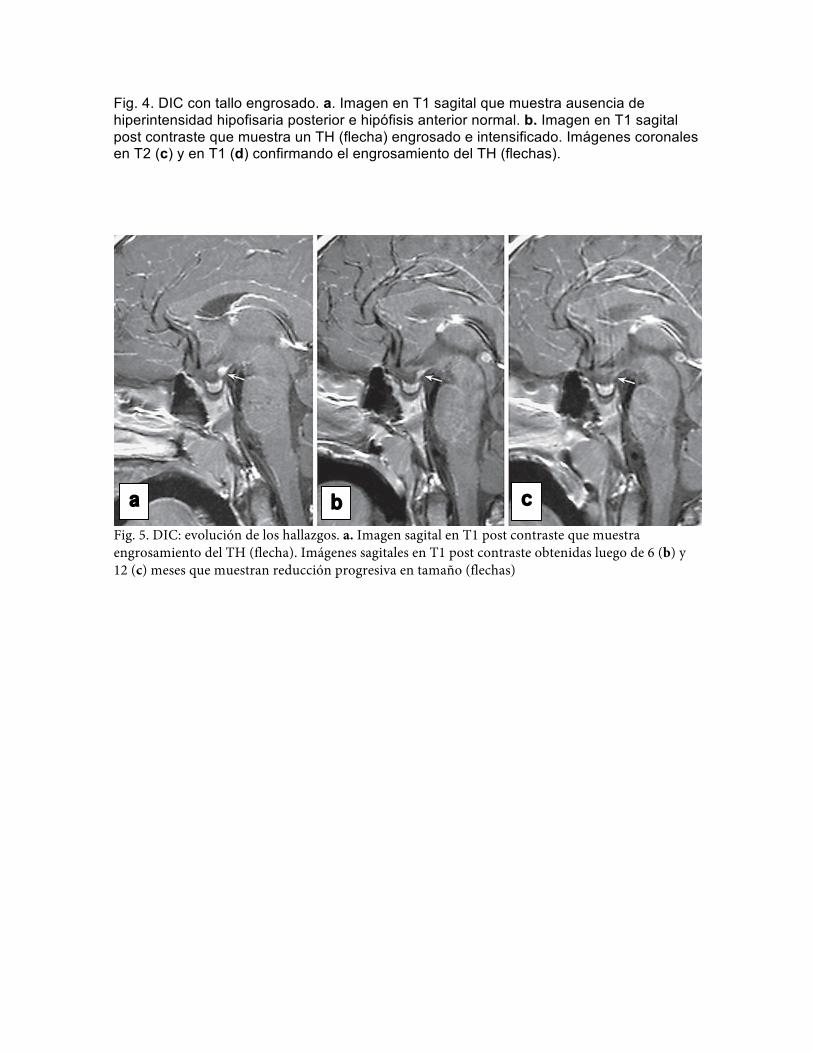
Fig. 4. DIC con tallo engrosado. a. Imagen en T1 sagital que muestra ausencia de hiperintensidad hipofisaria posterior e hipófisis anterior normal. b. Imagen en T1 sagital post contraste que muestra un TH (flecha) engrosado e intensificado. Imágenes coronales en T2 (c) y en T1 (d) confirmando el engrosamiento del TH (flechas).
Fig. 5. DIC: evolución de los hallazgos. a. Imagen sagital en T1 post contraste que muestra engrosamiento del TH (flecha). Imágenes sagitales en T1 post contraste obtenidas luego de 6 (b) y 12 (c) meses que muestran reducción progresiva en tamaño (flechas)
a b c
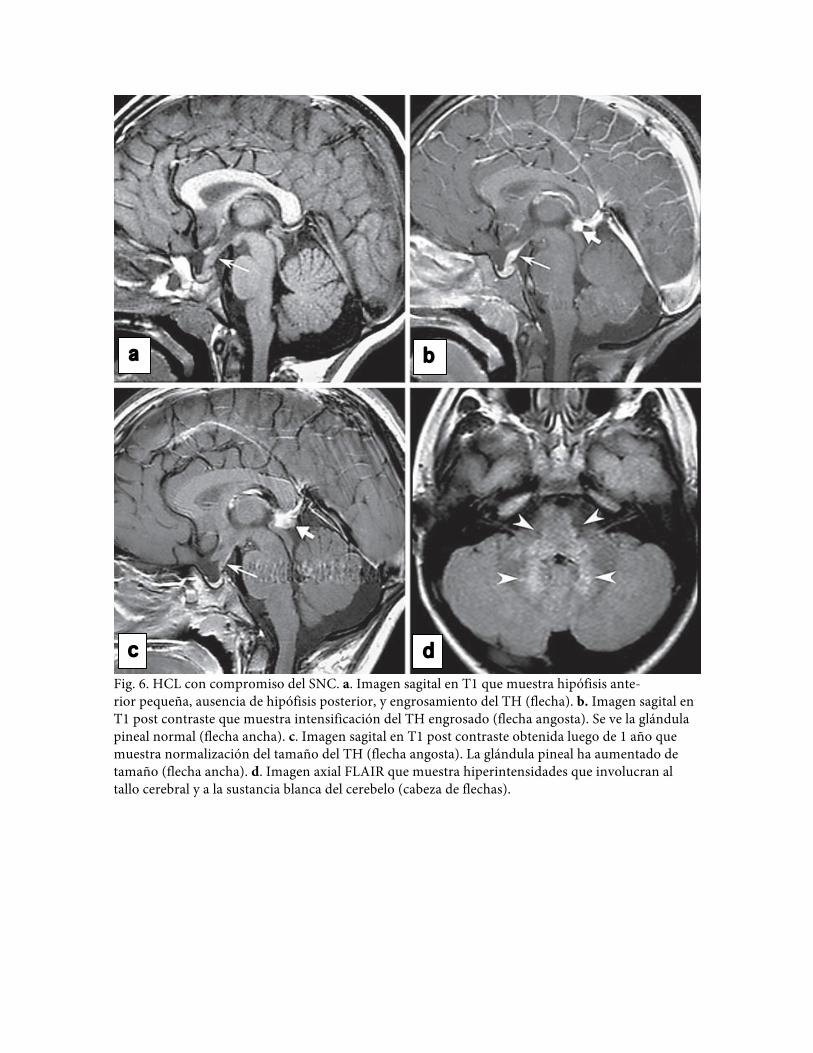
Fig. 6. HCL con compromiso del SNC. a. Imagen sagital en T1 que muestra hipófisis ante- rior pequeña, ausencia de hipófisis posterior, y engrosamiento del TH (flecha). b. Imagen sagital en T1 post contraste que muestra intensificación del TH engrosado (flecha angosta). Se ve la glándula pineal normal (flecha ancha). c. Imagen sagital en T1 post contraste obtenida luego de 1 año que muestra normalización del tamaño del TH (flecha angosta). La glándula pineal ha aumentado de tamaño (flecha ancha). d. Imagen axial FLAIR que muestra hiperintensidades que involucran al tallo cerebral y a la sustancia blanca del cerebelo (cabeza de flechas).
a b
c d

Fig. 7. Germinoma. Imágenes sagitales en T1 (a) y coronal en T2 (b) que muestran engrosamiento del tallo y de la glándula hipofisaria (flechas). El punto brillante posterior no está visible. El quiasma óptico está comprimido y desplazado hacia arriba (cabezas de flecha). c Imagen sagital en T1 post contraste que muestra al tejido patológico extenderse al lóbulo posterior de la hipófisis (cabeza de flechas). El lóbulo anterior está desplazado hacia adelante en la fosa hipofisaria y es visible como un área de mayor intensidad (flecha ancha).d. Imagen coronal en T1 post contraste confirma tejido patológico (flecha) que causa engrosamiento del tallo e invade la fosa pituitaria.
a b
c d
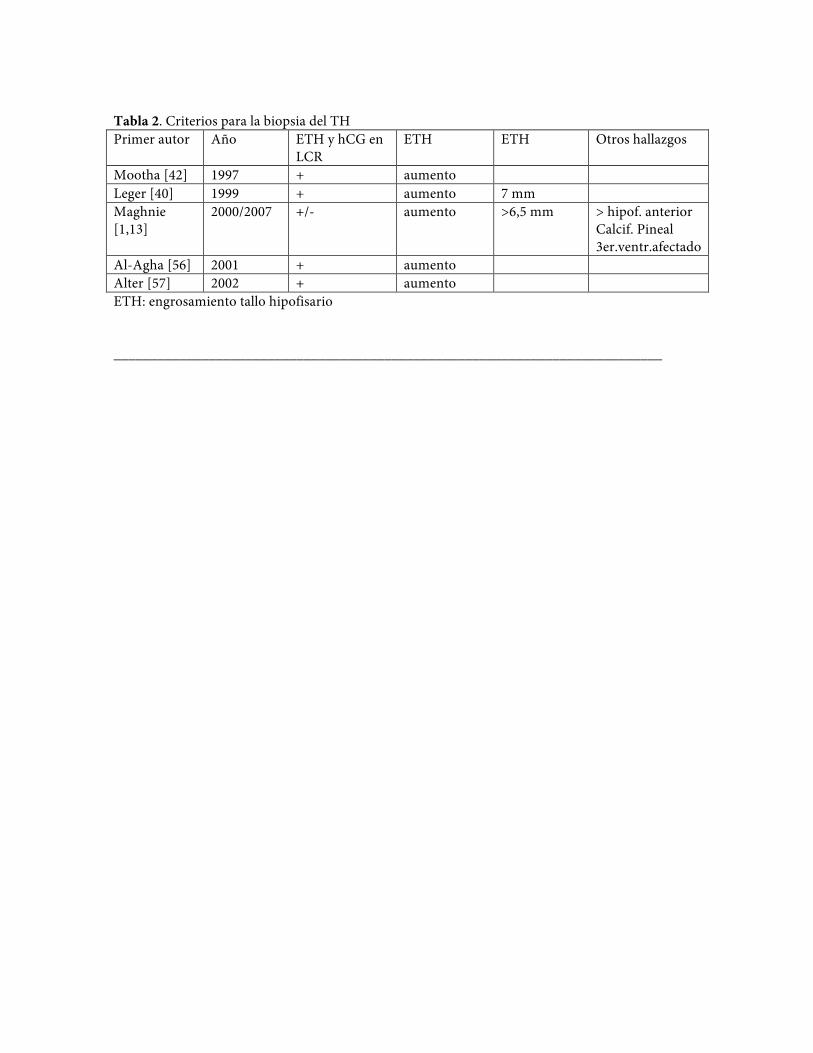
Tabla 2. Criterios para la biopsia del TH Primer autor Año ETH y hCG en
LCR ETH ETH Otros hallazgos
Mootha [42] 1997 + aumento Leger [40] 1999 + aumento 7 mm Maghnie [1,13]
2000/2007 +/- aumento >6,5 mm > hipof. anterior Calcif. Pineal 3er.ventr.afectado
Al-Agha [56] 2001 + aumento Alter [57] 2002 + aumento ETH: engrosamiento tallo hipofisario ___________________________________________________________________________
Si No � AVP/copeptinb normal Si No
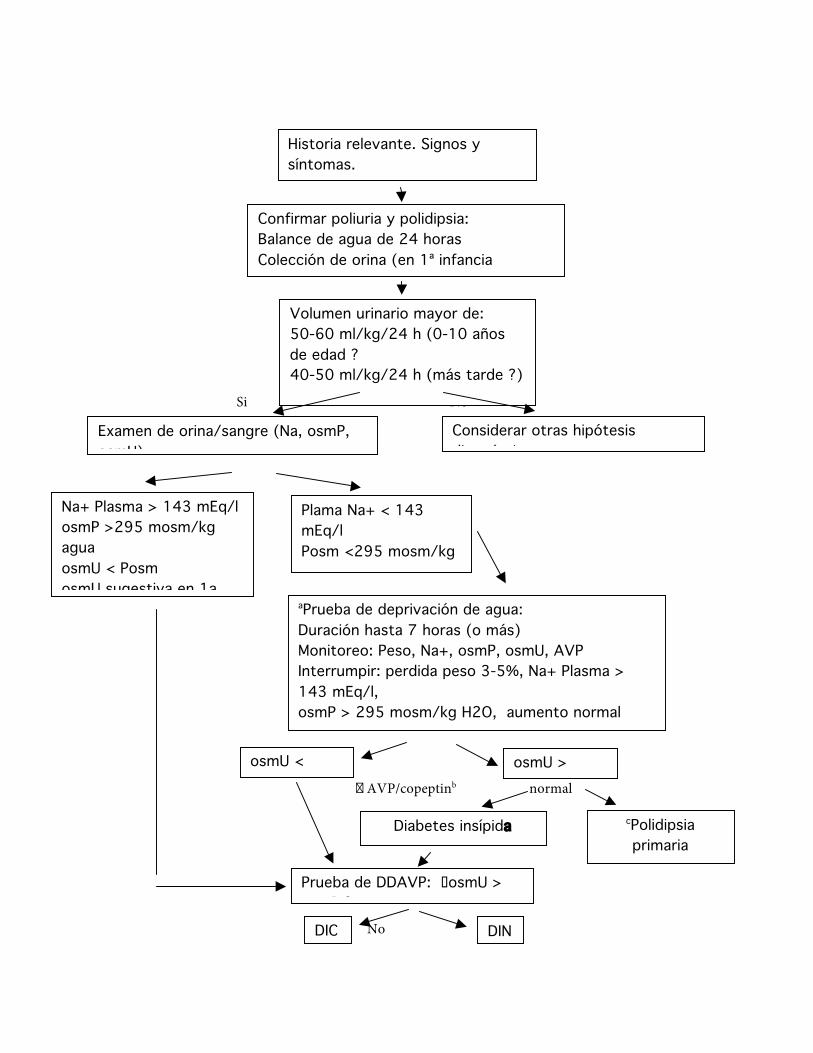
Historia relevante. Signos y síntomas. Examen clínico.
Confirmar poliuria y polidipsia: Balance de agua de 24 horas Colección de orina (en 1ª infancia cateterización) Examen clínico.
Volumen urinario mayor de: 50-60 ml/kg/24 h (0-10 años de edad ? 40-50 ml/kg/24 h (más tarde ?)
Examen de orina/sangre (Na, osmP, osmU)
Considerar otras hipótesis diagnósticas
aPrueba de deprivación de agua: Duración hasta 7 horas (o más) Monitoreo: Peso, Na+, osmP, osmU, AVP Interrumpir: perdida peso 3-5%, Na+ Plasma > 143 mEq/l, osmP > 295 mosm/kg H2O, aumento normal osmU, b AVP/copeptina al interrumpir cuando disponible osmU <
osmP osmU > osmP
Na+ Plasma > 143 mEq/l osmP >295 mosm/kg agua osmU < Posm osmU sugestiva en 1a infancia
Plama Na+ < 143 mEq/l Posm <295 mosm/kg agua Uosm < Posm
Diabetes insípida parcial
cPolidipsia primaria
Restricción de agua
Prueba de DDAVP: �osmU > osmP ?
DIC DIN

Fig. 8. Cuadro de flujo diagnóstico para la poliuria-polidipsia (relación normal osmU/osmP 1-1,5). osmP = osmolaridad plasmática; osmU = osmolaridad urinaria. a Laboratorio, b Confirmatorio especialmente en la diabetes insípida parcial, c La polidipsia primaria tiene una característica diferente de Na+ y osmP que no alcanza el cut-off de reportado.
__________________________________________________________________________________
Fig. 9. Esquema de flujo diagnóstico para la DIC. PLAP = Fosfatasa alcalina placentaria; SHHP = señal hiper-intensa de la hipófisis posterior (HP); AH = hipófisis anterior
Edad al comienzo, historia familiar, traumatismo de cerebro. Síntomas y signos de poliuria y polidipsia aparte: cefalea, alteraciones visuales, dolor óseo, convulsiones, signos neurológicos, erupciones, artritis, tos. Buscar signos clínicos de lesiones extracraneales: hueso, torax, piel, oído (otitis), ojos (uveítis), hígado (colestasis), nódulos linfáticos. Estudios endocrinológicos
RMN
Masa sugestiva Citologia del LCR, marcadores, pleiocitosis. Biopsia – confirmación histológica no necesaria en germinoma doble Terapia específica
Espesor del TH <6,5-7 mm SHHP ausente HA normal o reducida Déficits de HA +/-
*TH normal SHHP ausente/presente HA normal o descendida Déficit de HA +/-
RMN cada 4-6 meses por 2 años luego cada año por 3 años Estudio endocrino
Sintomas adicionales: Opciones: Recuperación espontánea Anti-AVPc (si disponible) Tecnecio 99m- RMN-STIR Rayo-X/TC torax Rayo-X hueso Biopsia extracraneal.
Progresión de la lesión (>6,5-7 mm) y/o del tamaño de la HA (germ. neurohipofisario) 3er. Ventrículo involucrado
Otras evoluciones. Recuperación espontánea Proceso autoimmune Anti-AVPc (si disponible) HCL Neurosarcoidosis Scan Tecnecio-99m RMN-STIR Biopsia-terapia específica Rayo-X/TC torax Seguimiento prolongado
Citología LCR, marcadores. Proteínas, pleiocitosis. Marcadores tumorales áreas selectas (hCG, PLAP, c-kit)
Tamaño normal de HP/HA Función de HA normal Anti-cAVP (si disponible) RMN dinámica Enfermedad vascular? Idiopática? Otra condición?
LCR negativo
LCR positivo
biopsia Terapia específica

La DIC y las anomalías de la sed Los trastornos adípsicos se caracterizan por una falta de sed inapropiada, con la falla de bebida para corregir la hiperosmolaridad. Se ha reportado recientemente que la incidencia de anomalías de la sed post-operatorias es de aproximadamente 1/3 de los pacientes con craneofaringeoma [59]. La DIC adipsica se caracteriza por escores anormalmente bajos de sed ausencia de respuesta de sed a la hipertonicidad marcada durante la infusión de solución salina. Los pacientes con craneofaringeoma que desarrollan un síndrome de adipsia y DIC post operatoria, fracasan en elevar la AVP del suero en respuesta a hipotensión inducida por drogas; más aún, no expresam sensación de sed ni luego de la hipotensión arterial ni luego de la infusión salina, indicando que están involucradas ambas vías, osmótica y no-osmótica [45]. La falla en la secreción de AVP en respuesta a hipotensión e hipovolemia podría aumentar el riesgo de deshidratación e hipernatremia con riesgo de vida. En los pacientes adípsicos debe establecerse una ingesta fija de fluídos, apropiada para el peso al cual se sabe que el paciente está eunatrémico y euvolémico. Debe administrarse DDAVP a una dosis y frecuencia capaz de conseguir una flujo de orina apropiado y un balance de líquidos equilibrado, calculando las pérdidas insensibles; es obligatorio el control adecuado del peso y de los niveles de sodio en suero. Aunque se sabe poco de cómo el cerebro orquesta los sistemas de osmoregulación, se han hecho recientemente avances nuestra comprensión de los mecanismos moleculares, celulares, y de las redes funcionales que median el control central de la homeostasis osmótica en los mamíferos [60]. A pesar del hecho de que los osmoreceptores cerebrales tienen un rol determinante en el control de las respuestas osmoreguladoras, los osmoreceptores periféricos también contribuyen al balance de fluídos. En efecto, pacientes con anormalidades complejas de la línea media del SNC, tales como la displasia septo-óptica principalmente tienen defectos en la osmoregulación de la AVP más que franca DIC [61], lo que sugiere que la disrupción de los mecanismos de los osmoreceptores contribuye significativamente a la etiología de este trastorno homeostático. Manejo Tratamiento de la DIC El medicamento de elección para el tratamiento de la diabetes insípida es la DDAVP, un análogo sintético de la hormona endógena AVP arginina, pero con un efecto vasopresor 2000 a 3000 veces menor. La DDAVP puede ser administrada por vía oral, intranasal o parenteral. Dada por vía intranasal u oral, las concentraciones máximas se alcanzan en 40-55 minutos. La vida media de la droga es de 3,5 horas. Generalmente, el flujo de orina disminuye 1 o 2 horas después de la administración y la duración de la acción varía entre 6 a 18 horas.

Hay una amplia variación individual en la dosis requerida para controlar la diuresis. Las dosis con las preparaciones orales (20 veces menos potente que la forma intranasal) varían desde 100 a 1200 µg, dividida en tres dosis, para la preparación intranasal aproximadamente entre 2-40 µg (una o dos veces por día), y para la parenteral 0,1-1 µg. Se debe iniciar con una dosis baja y aumentar según las necesidades. En la primera infancia una estrategia posible es dar solamente hidratación. Para el tratamiento de lactantes con DDAVP frecuentemente se administran dosis diluidas. Una alternativa segura es dejar pasar un tiempo corto de diuresis entre dos dosis. Debido a que la DDAVP se reduce cuando es diluida, estas preparaciones no deben guardarse por más de una semana. En niños de más edad, el spray intranasal y la forma oral son las formas más usadas en la DIC (5-20 µg una o dos veces por día). La DDAVP oral es especialmente conveniente en la niñez. Sus características positivas incluyen mejor absorción, menores complicaciones, y, debido a la fácil ruta de administración, buen cumplimiento de la medicación entre niños y adolescentes. La hiponatremia dilucional sintomática es el único riesgo potencial cuando la DDAVP se administra en exceso durante un período de tiempo prolongado. Los síntomas de hiponatremia incluyen cefalea, nausea, vómitos y convulsiones. Si no se tratan, estos síntomas pueden llevar al coma y a la muerte. Sin embargo, también puede ocurrir una hiponatremia asintomática. Debe tenerse especial cuidado en casos de terapias múltiples, por el riesgo de mielinosis extrapontina [62]. La administración por vía intranasal de DDAVP tiene efectos secundarios raros que incluyen irritación ocular, dolor de cabeza, mareos, rinitis o epistaxis, tos, enrojecimiento, nausas, vómitos, dolor abdominal, dolor de pecho, palpitaciones y taquicardia [63]. La evidencia disponible hoy es que el uso de DDAVP durante la gestación es seguro y no está relacionado con efectos adversos en la madre ni en el feto/niño [64]. En presencia de adipsia o hipodipsia, la diabetes insípida presenta un desafío dificultoso e inicialmente es mejor manejarse ajustando la dosis de DDAVP y la ingesta de fluidos en el ámbito de una internación hospitalaria. El peso diario puede ser usado como índice de balance de fluidos, pero también se requiere un monitoreo regular de los electrolitos.

Referencias 1 Maghnie M, Cosi G, Genovese E, Manca-Bitti ML, Cohen A, Zecca S, Tinelli C, Gallucci M, Bernasconi S, Boscherini B, Severi F, Arico M: Central diabetes insipidus in children and young adults. N Engl J Med 2000; 343:998–1007. 2 Hensen J, Buchfelder M: The posterior pituitary and its disease; in Pinchera A, et al (eds): Endocrinology and Metabolism. New York, McGraw-Hill, 2001, pp 99–115. 3 Fujiwara TM, Bichet DG: Molecular biology of hereditary diabetes insipidus. J Am Soc Nephrol 2005; 16: 2836–2846. 4 Bichet DG: Vasopressin receptor mutations in nephrogenic diabetes insipidus. Semin Nephrol 2008; 28: 245–251. 5 Robinson A, Verbalis J: Posterior pituitary; in Kronenberg H, et al (eds): Williams Textbook of Endocrinology, ed 11. Philadephia, Saunders Elsevier, 2008, pp263–287. 6 Christensen JH, Rittig S: Familial neurohypophyseal diabetes insipidus – an update. Semin Nephrol 2006; 26: 209–223. 7 Barat C, Simpson L, Breslow E: Properties of human vasopressin precursor constructs: inefficient monomer folding in the absence of copeptin as a potential contributor to diabetes insipidus. Biochemistry 2004; 43: 8191–8203. 8 De Mota N, Reaux-Le Goazigo A, El Messari S, Chartrel N, Roesch D, Dujardin C, Kordon C, Vaudry H, Moos F, Llorens-Cortes C: Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc Natl Acad Sci USA 2004; 101: 10464–10469. 9 Agre P: Nobel lecture. Aquaporin water channels. Biosci Rep 2004; 24: 127–163. 10 Engel A, Fujiyoshi Y, Agre P: The importance of aquaporin water channel protein structures. EMBO J 2000; 19: 800–806. 11 Robertson GL: Posterior pituitary; in Felig B, Frohman LA (eds): Endocrinology and Metabolism, ed 4. New York, McGraw Hill, 2001, p 234. 12 Braverman LE, Mancini JP, McGoldrick DM: Hereditary idiopathic diabetes insipidus: a case report with autopsy findings. Ann Intern Med 1965; 63: 503–508. 13 Ghirardello S, Garre ML, Rossi A, Maghnie M: The diagnosis of children with central diabetes insipidus. J Pediatr Endocrinol Metab 2007; 20: 359–375. 14 Willcutts MD, Felner E, White PC: Autosomal recessive familial neurohypophyseal diabetes insipidus with continued secretion of mutant weakly active vasopressin. Hum Mol Genet 1999; 8: 1303–1307. 15 Abu Libdeh A, Levy-Khademi F, Abdulhadi-Atwan M, Bosin E, Korner M, White PC, Zangen DH: Autosomal recessive familial neurohypophyseal diabetes insipidus: onset in early infancy. Eur J Endocrinol 2010; 162: 221–226. 16 Ye L, Li X, Chen Y, Sun H, Wang W, Su T, Jiang L, Cui B, Ning G: Autosomal dominant neurohypophyseal diabetes insipidus with linkage to chromosome 20p13 but without mutations in the AVP-NPII gene. J Clin Endocrinol Metab 2005; 90: 4388–4393. 17 Christensen JH, Siggaard C, Corydon TJ, Robertson GL, Gregersen N, Bolund L, Rittig S: Differential cellular handling of defective arginine vasopressin (AVP) prohormones in cells expressing mutations of the AVP gene associated with autosomal dominant and recessive familial neurohypophyseal diabetes insipidus. J Clin Endocrinol Metab 2004; 89: 4521–4531.

18 Davies J, Murphy D: Autophagy in hypothalamic neurones of rats expressing a familial neurohypophysial diabetes insipidus transgene. J Neuroendocrinol 2002; 14: 629–637. 19 Russell TA, Ito M, Yu RN, Martinson FA, Weiss J, Jameson JL: A murine model of autosomal dominant neurohypophyseal diabetes insipidus reveals progressive loss of vasopressin-producing neurons. J Clin Invest 2003; 112: 1697–1706. 20 Wahlstrom JT, Fowler MJ, Nicholson WE, Kovacs WJ: A novel mutation in the preprovasopressin gene identified in a kindred with autosomal dominant neurohypophyseal diabetes insipidus. J Clin Endocrinol Metab 2004; 89: 1963–1968. 21 Christensen JH, Siggaard C, Corydon TJ, Robertson GL, Gregersen N, Bolund L, Rittig S: Impaired trafficking of mutated AVP prohormone in cells expressing rare disease genes causing autosomal dominant familial neurohypophyseal diabetes insipidus. Clin Endocrinol 2004; 60: 125–136. 22 Ito M, Yu RN, Jameson JL: Mutant vasopressin precursors that cause autosomal dominant neurohypophyseal diabetes insipidus retain dimerization and impair the secretion of wild-type proteins. J Biol Chem 1999; 274: 9029–9037. 23 Friberg MA, Spiess M, Rutishauser J: Degradation of wild-type vasopressin precursor and pathogenic mutants by the proteasome. J Biol Chem 2004; 279: 19441–19447. 24 Maghnie M, Ghirardello S, De Bellis A, di Iorgi N, Ambrosini L, Secco A, De Amici M, Tinelli C, Bellastella A, Lorini R: Idiopathic central diabetes insipidus in children and young adults is commonly associated with vasopressin-cell antibodies and markers of autoimmunity. Clin Endocrinol 2006; 65: 470–478. 25 Pivonello R, De Bellis A, Faggiano A, Di Salle F, Petretta M, Di Somma C, Perrino S, Altucci P, Bizzarro A, Bellastella A, Lombardi G, Colao A: Central diabetes insipidus and autoimmunity: relationship between the occurrence of antibodies to arginine vasopressin- secreting cells and clinical, immunological, and radiological features in a large cohort of patients with central diabetes insipidus of known and unknown etiology. J Clin Endocrinol Metab 2003; 88: 1629–1636. 26 Bellastella G, Rotondi M, Pane E, Dello Iacovo A, Pirali B, Dalla Mora L, Falorni A, Sinisi AA, Bizzarro A, Colao A, Chiovato L, De Bellis A: Predictive role of the immunostaining pattern of immunofluorescence and the titers of antipituitary antibodies at presentation for the occurrence of autoimmune hypopituitarism in patients with autoimmune polyendocrine syndromes over a five year follow-up. J Clin Endocrinol Metab 2010; 95: 3750–3757. 27 Maghnie M, Genovese E, Sommaruga MG, Arico M, Locatelli D, Arbustini E, Pezzotta S, Severi F: Evolution of childhood central diabetes insipidus into panhypopituitarism with a large hypothalamic mass: is ‘lymphocytic infundibulo neurohypophysitis’ in children a different entity? Eur J Endocrinol 1998; 139: 635–640. 28 De Jersey J, Carmignac D, Le Tissier P, Barthlott T, Robinson I, Stockinger B: Factors affecting the susceptibility of the mouse pituitary gland to CD8 T-cell-mediated autoimmunity. Immunology 2004; 111: 254–261. 29 Tzou SC, Lupi I, Landek M, Gutenberg A, Tzou YM, Kimura H, Pinna G, Rose NR, Caturegli P: Autoimmune hypophysitis of SJL mice: clinical insights from a new animal model. Endocrinology 2008; 149: 3461–3469. 30 Imura H, Nakao K, Shimatsu A, Ogawa Y, Sando T, Fujisawa I, Yamabe H: Lymphocytic infundibulo neurohypophysitis as a cause of central diabetes insipidus. N Engl J Med 1993; 329: 683–689. 31 Mirocha S, Elagin RB, Salamat S, Jaume JC: T regulatory cells distinguish two types of primary hypophysitis. Clin Exp Immunol 2009; 155: 403–411. 32 Marchand I, Barkaoui MA, Garel C, Polak M, Donadieu J: Central diabetes insipidus as the inaugural manifestation of Langerhans cell histiocytosis: natural history and medical evaluation of 26 children and adolescents. J Clin Endocrinol Metab 2011; 96:1352–1360.

33 Rutishauser J, Kopp P, Gaskill MB, Kotlar TJ, Robertson GL: Clinical and molecular analysis of three families with autosomal dominant neurohypophyseal diabetes insipidus associated with a novel and recurrent mutations in the vasopressin-neurophysin II gene. Eur J Endocrinol 2002; 146: 649–656. 34 Batista SL, Moreira AC, Antunes-Rodrigues J, Castro M, Elias LL, Elias PC: Clinical features and molecular analysis of arginine-vasopressin neurophysin II gene in long-term follow-up patients with idiopathic central diabetes insipidus. Arq Bras Endocrinol Metabol 2010; 54: 269–273. 35 Maghnie M, Altobelli M, Di Iorgi N, Genovese E, Meloni G, Manca-Bitti ML, Cohen A, Bernasconi S: Idiopathic central diabetes insipidus is associated with abnormal blood supply to the posterior pituitary gland caused by vascular impairment of the inferior hypophyseal artery system. J Clin Endocrinol Metab 2004; 89: 1891–1896. 36 Grois N, Potschger U, Prosch H, Minkov M, Arico M, Braier J, Henter JI, Janka-Schaub G, Ladisch S, Ritter J, Steiner M, Unger E, Gadner H: Risk factors for diabetes insipidus in Langerhans cell histiocytosis. Pediatr Blood Cancer 2006; 46: 228–233. 37 Maghnie M, Bossi G, Klersy C, Cosi G, Genovese E, Arico M: Dynamic endocrine testing and magnetic resonance imaging in the long-term follow-up of childhood Langerhans cell histiocytosis. J Clin Endocrinol Metab 1998; 83: 3089–3094. 38 Donadieu J, Rolon MA, Pion I, Thomas C, Doz F, Barkaoui M, Robert A, Deville A, Mazingue F, David M, Brauner R, Cabrol S, Garel C, Polak M: Incidence of growth hormone deficiency in pediatric-onset Langerhans cell histiocytosis: efficacy and safety of growth hormone treatment. J Clin Endocrinol Metab 2004; 89: 604–609. 39 Maghnie M, Villa A, Arico M, Larizza D, Pezzotta S, Beluffi G, Genovese E, Severi F: Correlation between magnetic resonance imaging of posterior pituitary and neurohypophyseal function in children with diabetes insipidus. J Clin Endocrinol Metab 1992; 74: 795–800. 40 Leger J, Velasquez A, Garel C, Hassan M, Czernichow P: Thickened pituitary stalk on magnetic resonance imaging in children with central diabetes insipidus. J Clin Endocrinol Metab 1999; 84: 1954–1960. 41 Ghirardello S, Malattia C, Scagnelli P, Maghnie M: Current perspective on the pathogenesis of central diabetes insipidus. J Pediatr Endocrinol Metab 2005; 18: 631–645. 42 Mootha SL, Barkovich AJ, Grumbach MM, Edwards MS, Gitelman SE, Kaplan SL, Conte FA: Idiopathic hypothalamic diabetes insipidus, pituitary stalk thickening, and the occult intracranial germinoma in children and adolescents. J Clin Endocrinol Metab 1997; 82: 1362–1367. 43 Bettendorf M, Fehn M, Grulich-Henn J, Selle B, Darge K, Ludecke DK, Heinrich UE, Saeger W: Lymphocytic hypophysitis with central diabetes insipidus and consequent panhypopituitarism preceding a multifocal, intracranial germinoma in a prepubertal girl. Eur J Pediatr 1999; 158: 288–292. 44 Prosch H, Grois N, Bokkerink J, Prayer D, Leuschner I, Minkov M, Gadner H: Central diabetes insipidus: Is it Langerhans cell histiocytosis of the pituitary stalk? A diagnostic pitfall. Pediatr Blood Cancer 2006; 46: 363–366. 45 Ghirardello S, Hopper N, Albanese A, Maghnie M: Diabetes insipidus in craniopharyngioma: postoperative management of water and electrolyte disorders. J Pediatr Endocrinol Metab 2006; 19 (suppl 1):413–421. 46 Finken MJ, Zwaveling-Soonawala N, Walenkamp MJ, Vulsma T, van Trotsenburg AS, Rotteveel J: Frequent occurrence of the triphasic response (diabetes insipidus/hyponatremia/diabetes insipidus) after surgery for craniopharyngioma in childhood. Horm Res Paediatr 2011; 76: 22–26. 47 Repaske DR, Medlej R, Gultekin EK, Krishnamani MR, Halaby G, Findling JW, Phillips

JA 3rd: Heterogeneity in clinical manifestation of autosomal dominant neurohypophyseal diabetes insipidus caused by a mutation encoding Ala-1– 1 Val in the signal peptide of the arginine vasopressin/neurophysin II/copeptin precursor. J Clin Endocrinol Metab 1997; 82: 51–56. 48 Barrett TG, Bundey SE, Macleod AF: Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995; 346: 1458–1463. 49 Giuliano F, Bannwarth S, Monnot S, Cano A, Chabrol B, Vialettes B, Delobel B, Paquis- Flucklinger V: Wolfram syndrome in French population: characterization of novel mutations and polymorphisms in the WFS1 gene. Hum Mutat 2005; 25: 99–100. 50 Smith CJ, Crock PA, King BR, Meldrum CJ, Scott RJ: Phenotype-genotype correlations in a series of Wolfram syndrome families. Diabetes Care 2004; 27: 2003–2009. 51 Medlej R, Wasson J, Baz P, Azar S, Salti I, Loiselet J, Permutt A, Halaby G: Diabetes mellitus and optic atrophy: a study of Wolfram syndrome in the Lebanese population. J Clin Endocrinol Metab 2004; 89: 1656–1661. 52 Baylis PH, Cheetham T: Diabetes insipidus. Arch Dis Child 1998; 79: 84–89. Kanno K, Sasaki S, Hirata Y, Ishikawa S, Fushimi K, Nakanishi S, Bichet DG, Marumo F: Urinary excretion of aquaporin-2 in patients with diabetes insipidus. N Engl J Med 1995; 332: 1540–1545. 54 Morgenthaler NG, Struck J, Jochberger S, Dunser MW: Copeptin: clinical use of a new biomarker. Trends Endocrinol Metab 2008; 19: 43–49. 55 Maghnie M, Genovese E, Bernasconi S, Binda S, Arico M: Persistent high MR signal of the posterior pituitary gland in central diabetes insipidus. AJNR Am J Neuroradiol 1997; 18: 1749–1752. 56 Al-Agha AE, Thomsett MJ, Ratcliffe JF, Cotterill AM, Batch JA: Acquired central diabetes insipidus in children: a 12-year Brisbane experience. J Paediatr Child Health 2001; 37:172–175. 57 Alter CA, Bilaniuk LT: Utility of magnetic resonance imaging in the evaluation of the child with central diabetes insipidus. J Pediatr Endocrinol Metab 2002; 15(suppl 2):681– 687. 58 Maghnie M, Genovese E, Arico M, Villa A, Beluffi G, Campani R, Severi F: Evolving pituitary hormone deficiency is associated with pituitary vasculopathy: dynamic MR study in children with hypopituitarism, diabetes insipidus, and Langerhans cell histiocytosis. Radiology 1994; 193: 493–499. 59 Smith D, McKenna K, Moore K, Tormey W, Finucane J, Phillips J, Baylis P, Thompson CJ: Baroregulation of vasopressin release in adipsic diabetes insipidus. J Clin Endocrinol Metab 2002; 87: 4564–4568. 60 Bourque CW: Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci 2008; 9: 519–531. 61 Secco A, Allegri AE, di Iorgi N, Napoli F, Calcagno A, Bertelli E, Olivieri I, Pala G, Parodi S, Gastaldi R, Rossi A, Maghnie M: Posterior pituitary (PP) evaluation in patients with anterior pituitary defect associated with ectopic PP and septo-optic dysplasia. Eur J Endocrinol 2011; 165: 411–420. 62 Maghnie M, Genovese E, Lundin S, Bonetti F, Arico M: Iatrogenic [corrected] extrapontine myelinolysis in central diabetes insipidus: are cyclosporine and 1-desamino-8-Darginine vasopressin harmful in association? J Clin Endocrinol Metab 1997; 82: 1749–1751. 63 Vande Walle J, Stockner M, Raes A, Norgaard JP: Desmopressin 30 years in clinical use: a safety review. Curr Drug Saf 2007; 2:232–238. 64 Kim RJ, Malattia C, Allen M, Moshang T Jr, Maghnie M: Vasopressin and desmopressin in central diabetes insipidus: adverse effects and clinical considerations. Pediatr Endocrinol Rev 2004; 2 (suppl 1):115–123.



















