
HIPERINSULINISMO CONGÉNITO E HIPOGLUCEMIA NEONATAL.
55
Causas de la Hipoglucemia Neonatal Diagnóstico y tratamiento del Hiperinsulinismo Congénito (Agosto 2013) Dr. Eduardo R. Guzmán Lic. Elizabeth K. Malament
-
Upload
hicongenito -
Category
Documents
-
view
5.703 -
download
0
Transcript of HIPERINSULINISMO CONGÉNITO E HIPOGLUCEMIA NEONATAL.

Causas de la Hipoglucemia Neonatal Diagnóstico y tratamiento del Hiperinsulinismo
Congénito(Agosto 2013)
Dr. Eduardo R. GuzmánLic. Elizabeth K. Malament

El Dr. René Favaloro solía decir: “La Medicina sin humanismo médico nomerecer ser ejercida”
Ser médico es mucho más que tener conocimientos científicos. Un científicosin humanidad puede ser fácilmente un “bárbaro ilustrado”.
Antes de ser buen médico, sé buena persona…
Si los bebés y niños que padecen Hiperinsulinismo Congénito, pudieran
expresarse, sin dudas, le pedirían a los médicos:
“Por favor, ayuda a salvar mi vida, a salvar mi páncreas, y a salvar mi futuro”

Bibliografía
Neonatal Hypoglycemia. Ved Bhushan Arya, Senthyl Senniappan, MariaGuemes, Khalid Hussain. Indian J. Pediatr. 2013
Hyperinsulinaemic Hypoglycaemia: Genetic Mechanisms, Diagnosis andManagement. Zainaba Mohamed, Ved Bhushan Arya, Khalid Hussain. J.Clin ResPediatr Endocrinol 2012; 4 (4): 169-181
Monogenic hyperinsunemic hypoglycemia: current insights into thepathogenesis and management. Katherine Lord, Diva D. De León. InternationalJournal of Pediatric Endocrinology 2013, 2013:3
The genetic basis of Congenital Hyperinsulinism. C James, R R Kapoor, DIsmail, et al. J. Med Genet 2009 46: 289-299
Congenital hyperinsulinism. Patrick A. Dillon. Curr Opin Pediatr. 2013 Jun; 25(3): 357-61
TABLAS Y FIGURAS
Tabla 1. Infantes con riesgo de hipoglucemia (Diapositiva 8)
Figura 1. Principales sitios reguladores de la secreción de insulina en lacélula β del páncreas (Diapositiva 13)
Figura 2. Estructura de los canales de potasio dependientes del ATP (KATP)(Diapositiva 14)
Figura 3. Etiología genética de una lesión focal (Diapositiva 17)
Tabla 2. Puntos claves a tener en cuenta en el Hiperinsulinismo Congénito(Diapositiva 22)
Figura 4. Esquema ilustrativo de la oxidación de los ácidos grasos(Diapositiva 28)

TABLAS Y FIGURAS
Figura 5. Esquema ilustrativo del trastorno en la oxidación de los ácidosgrasos (Diapositiva 30)
Tabla 3. Principales enfermedades de depósito de glucógeno (Diapositiva33)
Figura 6. Algoritmo para la interpretación de los resultados clínicos ybioquímicos de neonatos con hipoglucemia (Diapositiva 42)
Tabla 4: Criterios para el diagnóstico de Hiperinsulinismo Congénito(Diapositiva 46)
Figura 7: Algoritmo del enfoque terapéutico para el HiperinsulinismoCongénito (Diapositiva 50)

La glucosa es el combustible esencial para el metabolismo cerebral. Enconsecuencia, los niveles bajos de glucosa en sangre, pueden ocasionarencefalopatía.
La hipoglucemia neonatal puede ser transitoria (que se observacomúnmente en lactantes de alto riesgo) o permanente.
Existe una amplia gama de desórdenes endocrinológicos y metabólicosraros que pueden provocar hipoglucemia neonatal.
El Hiperinsulinismo Congénito (HIC) es responsable de la forma más gravede hipoglucemia.
El diagnóstico rápido con una intervención temprana agresiva es la base del tratamiento parar evitar el daño neurológico irreversible.
HIPOGLUCEMIA NEONATAL

Si bien no hay un consenso entre los expertos para definir el valor dehipoglucemia neonatal, sí se han determinado límites de riesgo para suintervención.
Para cualquier bebé con signos y síntomas compatibles conhipoglucemia, ésta debe mantenerse por encima de 45 mg/dl, A MENOS QUEHAYA UNA SOSPECHA DE HIPERINSULINISMO CONGÉNITO, EN LA QUE DEBEEVITARSE GLUCEMIAS INFERIORES A 65 mg/dl.
La mayoría de los casos de hipoglucemia neonatal son debido a la demora delos procesos normales de la adaptación metabólica después del nacimiento, yse producen en niños en riesgo (hipoglucemia neonatal transitoria).
Una etiología metabólica u hormonal subyacente debe sospecharse cuandola hipoglucemia es de severidad inusual o se produce en un bebé de bajoriesgo.

Tabla 1. Infantes con riesgo de hipoglucemia

HIPOGLUCEMIA NEONATAL PERSISTENTE
1. HIPERINSULINISMO CONGÉNITO
2. OTRAS CAUSAS ENDOCRINOLÓGICAS
3. DESÓRDENES METABÓLICOSa) Trastornos de la oxidación de ácidos grasosb) Trastornos del almacenamiento del glucógenoc) Trastornos de la gluconeogénesis

1. HIPERINSULINISMO CONGÉNITO
Es la causa más común de hipoglucemia recurrente y persistente en el reciénnacido.
Se caracteriza por la secreción no regulada y apropiada de insulina por las célulasβ del páncreas en presencia de una concentración baja de glucosa.
La detección temprana, el diagnóstico y el tratamiento inmediato sonfundamentales para evitar el daño neurológico.
El Hiperinsulinismo Congénito es una patología heterogénea en cuanto a lapresentación clínica, pero también desde el punto de vista genético e histológico.
Es uno de los trastornos más complicados y desafiantes que enfrentan losendocrinólogos pediátricos.
La necesidad de prevenir el daño neurológico permanente, hace que seafundamental identificar y tratar a estos niños tempranamente.

En la última década ha habido un enorme progreso en la comprensión de lasbases genéticas y moleculares del Hiperinsulinismo Congénito. Estacomprensión ha permitido avanzar en el tratamiento, y en mejoresresultados, en particular mediante el uso de la Tomografía por Emisión dePositrones con 18-fluoro-L-3,4 dihidroxifenilalanina para identificar y curarlesiones focales.
Hasta el momento se han identificado mutaciones en 9 genes(ABCC8, KCNJ11, GLUD1, GCK, HADHSC, SLC16A1, HNF4A, UCP2 Y HNF1A)involucrados en la patogénesis del HIC.

Hasta la actualidad se han identificado nueve genes involucrados en la patogénesis del HIC:
• Hiperinsulinismo asociado a defectos en el gen ABCC8 que codifican para SUR1 - receptor 1 de sulfunilurea-
• Hiperinsulinismo asociado a defectos en el KCNJ11 que codifica para Kir6.2 rectificador interno de los canales de potasio
• Hiperinsulinismo asociado a defectos en el gen GLUD1 que codifica para la enzima GDH –glutamato deshidrogenasa- (HI/HA Hiperinsulinismo/Hiperamonemia)
• Hiperinsulinismo asociado a defectos en el gen GCK que codifica para GK -Glucoquinasa-
• Hiperinsulinismo asociado a defectos en el gen HADHSC que codifica para SCHAD (3-hidroxi-acil-coA deshidrogenasa de cadena corta)
• Hiperinsulinismo asociado a defectos en el gen HNF4A que codifica para HNF-4α -factor nuclear 4 alfa de hepatocito-
• Hiperinsulinismo asociada a defectos en el gen HNF1A que codifica para HNF-1 α –factor nuclear 1 alfa de hepatocito-
• Hiperinsulinismo asociado a defectos en el gen UCP2 que codifica para la proteína desacoplante mitocondrial 2
• Hiperinsulinismo asociado a defectos en el gen SLC16A1 que codifica para el transportador 1 de Monocarboxilato

Figura 1. Principales sitios reguladores de la secreción de insulina en la célula β del páncreas. Cuando losniveles de glucosa aumentan, ésta ingresa a la célula a través del transportador específico GLUT-2, luego laglucosa es fosforilada por la enzima Glucokinasa (GK), aumentando así los niveles de ATP. Los niveles de ATPpueden verse incrementados por el metabolismo de otros sustratos, como los aminoácidos entre ellos elglutamato, vía glutamato deshidrogenasa. El incremento ATP/ADP causa el cierre de los canales KATP, lo queproduce la despolarización de la membrana celular, la apertura de los canales de calcio dependientes devoltaje, el aumento de la concentración de calcio intracelular, y finalmente, la secreción de insulina. Los genesinvolucrados en la patogénesis: ABCC8 (que codifica para SUR.1), KCNJ11 (que codifica para Kir6.2), GLUD1(que codifica para GDH), GCK (que codifica para GK), HADHSC (que codifica para SCHAD), HNF4A (quecodifica para HNF-4α), HNF1A (que codifica para HNF-1 α), UCP2 (que codifica para la proteína desacoplantemitocondrial 2) y SLC16A1 (que codifica para MCT1).
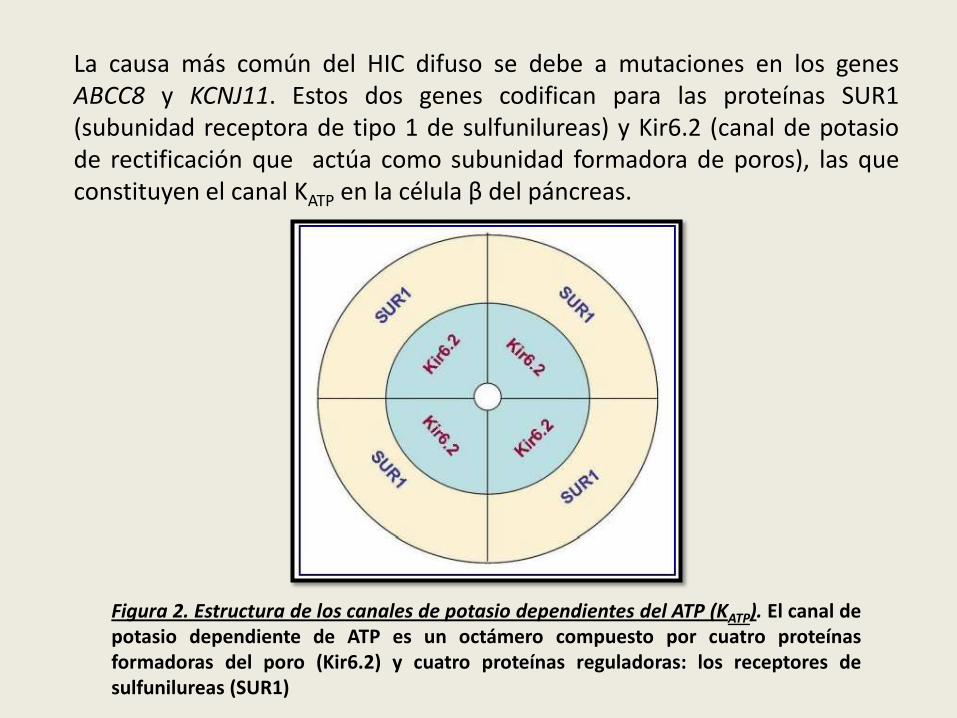
La causa más común del HIC difuso se debe a mutaciones en los genesABCC8 y KCNJ11. Estos dos genes codifican para las proteínas SUR1(subunidad receptora de tipo 1 de sulfunilureas) y Kir6.2 (canal de potasiode rectificación que actúa como subunidad formadora de poros), las queconstituyen el canal KATP en la célula β del páncreas.
Figura 2. Estructura de los canales de potasio dependientes del ATP (KATP). El canal depotasio dependiente de ATP es un octámero compuesto por cuatro proteínasformadoras del poro (Kir6.2) y cuatro proteínas reguladoras: los receptores desulfunilureas (SUR1)

La mayor parte de las afecciones genéticas se hallan en los genes ABCC8 yKCNJ11 (40-45% de los casos).
Las mutaciones inactivantes recesivas en ABCC8 y KCNJ11 causanusualmente HIC severo, el que no responde al tratamiento médico conDiazoxide.
Las mutaciones inactivantes dominantes en ABCC8 y KCNJ11 usualmentecausan un fenotipo más leve.
Otras mutaciones genéticas identificadas representan entre el 5-10 % delos casos, y resultan de anomalías enzimáticas o de anomalías en latranscripción.
La afecciones genéticas que producen HIC en el 45-55 % de los casosrestantes, es aún desconocida.

Hay dos tipos histológicos de HIC: focal y difuso.
En la forma difusa, todo el páncreas se ve afectado, mientras que en laforma focal, sólo una parte del páncreas está afectada. La forma difusa sehereda de forma autosómica recesiva o dominante, mientras que la formafocal se hereda por vía paterna combinada con una pérdida somática deheterocigosidad (LOH).
Estos pacientes heredan en forma recesiva una mutación paterna en el genABCC8 o KCNJ11. Durante el desarrollo pancreático, una única célulaprecursora pierde el alelo materno del cromosoma 11p15, de modo que laprogenie de esta célula sólo tendrá genes mutantes SUR1, y ademáscarecerá de los genes supresores del crecimiento impresos en el alelomaterno perdido (LOH). El resultado es una lesión focal proliferantecompuesta de células β que secretan insulina en forma desregulada.
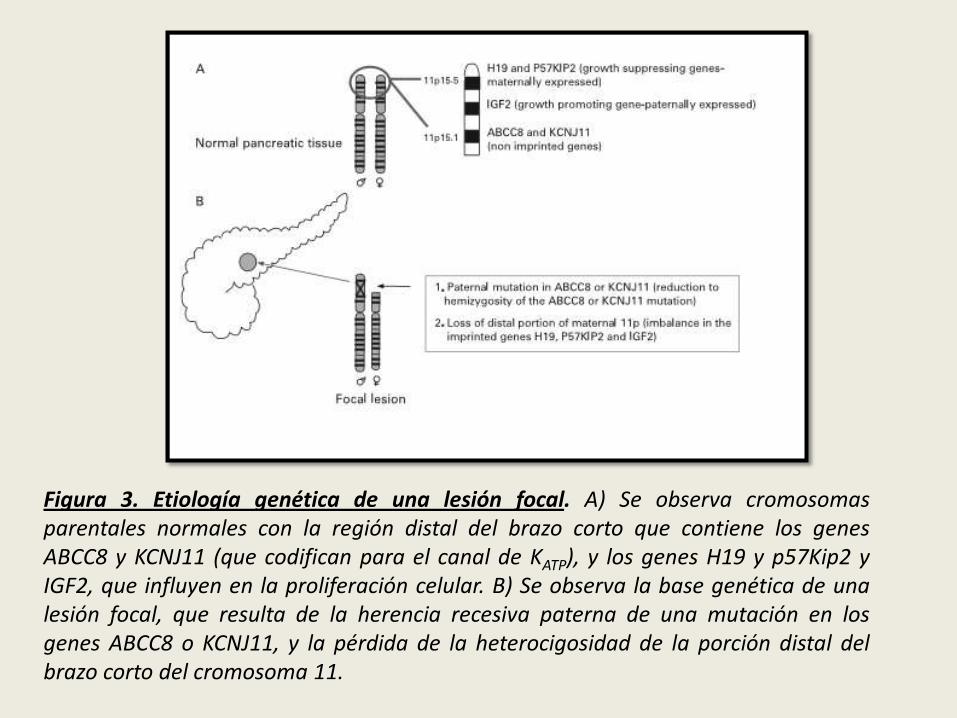
Figura 3. Etiología genética de una lesión focal. A) Se observa cromosomasparentales normales con la región distal del brazo corto que contiene los genesABCC8 y KCNJ11 (que codifican para el canal de KATP), y los genes H19 y p57Kip2 yIGF2, que influyen en la proliferación celular. B) Se observa la base genética de unalesión focal, que resulta de la herencia recesiva paterna de una mutación en losgenes ABCC8 o KCNJ11, y la pérdida de la heterocigosidad de la porción distal delbrazo corto del cromosoma 11.

Las lesiones focales son localizadas mediante Tomografía por Emisiónde Positrones con 18-fluoro-L-3,4 dihidroxifenilalanina, para luego serresecadas, logrando la curación del niño.
Las formas difusas de HIC que no responden al tratamientomédico, requieren pancreatectomía casi total (con el riesgoconsecuente de la Diabetes Mellitus y la insuficiencia pancreáticaexócrina).

El Síndrome de Hiperinsulinismo/Hiperamonemia (HI/HA), es la segundaforma más común de HIC asociada a mutaciones missense activantes en elgen GLUD1, el cual codifica para la enzima de la matriz mitocondrialglutamato deshidrogenasa (GDH).
Los pacientes con esta afección presentan hipoglucemia postprandialrecurrente sintomática luego de las comidas ricas en proteínas(hipoglucemia sensible a la leucina), así como hipoglucemia durante elayuno acompañada por elevaciones asintomáticas de amonio en plasma.
Los pacientes con Síndrome HI/HA presentan más complicacionesneurológicas como epilepsia y problemas de aprendizaje. El dosaje rutinariode concentraciones plasmáticas de amonio en todos los pacientes conhipoglucemia, constituye una prueba de detección esencial para estetrastorno.

Las mutaciones en los genes HNF4A (codifica para el Factor deTranscripción Nuclear de Hepatocito 4α), HNF1A (codifica para el Factorde Transcripción Nuclear de Hepatocito 1α) y GCK (codifica para la enzimaGlucoquinasa) que causan diabetes del adulto de inicio juvenil(MODY), también están involucradas en la génesis del HIC.
La gravedad del HIC en estos pacientes es variable ya que la hipoglucemiapuede ser controlada con dieta o ser persistente requiriendo Diazoxide, einclusive requerir cirugía en algunos pacientes con mutación en el genGCK.
La hipoglucemia hiperinsulinémica también pude estar asociada asíndromes de desarrollo como el Síndrome de Beckwith-Wiedemann ocondiciones metabólicas raras como desórdenes de glicosilación.

Los pacientes con HIC presentan mayor riesgo neurológico debido a que lasecreción desregulada de insulina conduce la glucosa dentro del músculoesquelético, el tejido adiposo e hígado, causando hipoglucemia severa.También dicha secreción desregulada de insulina inhibe la producción deglucosa a través de la glucolisis y la gluconeogénesis.Adicionalmente, inhibe la liberación de ácidos grasos y la síntesis decuerpos cetónicos, privando al cerebro tanto de glucosa como decombustibles alternativos.

Tabla 2. Puntos claves a tener en cuenta en el Hiperinsulinismo Congénito

2. OTRAS CAUSAS ENDOCRINOLÓGICAS DE LA HIPOGLUCEMIA NEONATAL
El sistema de contra-regulación hormonal garantiza un suministro continuode la glucosa a los órganos vitales.
El glucagón y la adrenalina son las hormonas más importantes para elrestablecimiento inmediato de la concentración de glucosa ensangre, mientras que el cortisol y la hormona de crecimiento ejercen supapel contrarregulador pasadas unas horas de comenzada la hipoglucemia.
La deficiencia de cualquiera de estas hormonas puede causar hipoglucemia.Sin embargo, no se ha descripto en ningún ser humano deficiencia genéticade glucagón o adrenalina.
El hipopituitarismo congénito puede causar hipoglucemia potencialmentemortal, concentraciones séricas de sodioanormales, shock, microfalo, ictericia y luego retraso del crecimiento.

El hipotiroidismo congénito ha sido reportado en un paciente conhipoglucemia neonatal persistente, pero el mecanismo no está claro.
La insuficiencia suprarrenal primaria o secundaria es una causa importantede la hipoglucemia en el recién nacido.
2. OTRAS CAUSAS ENDOCRINOLÓGICAS DE LA HIPOGLUCEMIA NEONATAL

3. DESÓRDENES METABÓLICOSa) Trastornos de la oxidación de ácidos grasosb) Trastornos del almacenamiento del glucógenoc) Trastornos de la gluconeogénesis
Los trastornos metabólicos incluyen: defectos en la β oxidación de ácidos
grasos, glucogenolisis, gluconeogénesis, metabolismo de la
carnitina, metabolismo de aminoácidos, síntesis y utilización de cuerpos
cetónicos, acidemias orgánicas, y trastornos de la cadena respiratoria
mitocondrial.

3. DESÓRDENES METABÓLICOSa) Trastornos de la oxidación de ácidos grasos
Las deficiencias de la oxidación de ácidos grasos son un grupo de
enfermedades genéticas, de carácter autosómico recesivo, cuya
principal característica clínica es la hipoglucemia hipocétosica asociada
con el ayuno; sin embargo, el espectro de síntomas clínicos es muy
amplio y abarca desde pacientes asintomáticos o con una leve hipotonía
hasta pacientes con debilidad muscular, cardiomiopatía y fallo hepático
o multisistémico.
En general, el pronóstico depende de la forma clínica de cada
paciente, pero en todos los casos, está condicionada a que se establezca
un diagnóstico temprano y un manejo terapéutico correcto.

3. DESÓRDENES METABÓLICOSa) Trastornos de la oxidación de ácidos grasos
Los ácidos grasos juegan un papel esencial en los momentos en que se requiera
suplir de forma eficaz la energía suministrada por la glucosa, esto es, durante
los períodos de ayuno prolongado o en momentos de gran requerimiento
energético (ejercicio prolongado, infecciones).
En estas situaciones, la oxidación de los ácidos grasos proporcionará los
elementos necesarios para que se realice de forma adecuada la
gluconeogénesis, la ureagenésis y la cetogénesis.

Figura 4. Esquema ilustrativo del mecanismo de la oxidación de los ácidos grasos. Los ácidosgrasos son una fuente directa de energía tanto para el músculo esquelético como para el músculocardíaco. En estos tejidos, el producto final de la β-oxidación (acetil-CoA) se dirigepreferentemente a la formación de C2O y H2O a través del ciclo de Krebs. En el hígado, el acetil-CoA producido se utiliza mayoritariamente para la formación de cuerpos cetónicos, que sonexportados a otros tejidos como combustible auxiliar. De esta forma, en los períodos de ayunoprolongado, el cerebro utiliza los cuerpos cetónicos producidos por la oxidación hepática de lasgrasas, como principal fuente energética.

Cuando está interrumpida la β-oxidación se produce una falta de producto final
(acetil-CoA), necesario para la formación de cuerpos cetónicos y para activar la
gluconeogénesis y la ureagenésis.
En consecuencia, ante los períodos de ayuno no se obtiene una respuesta
energética adecuada, produciéndose una hipoglucemia
hipocetósica, hiperamonemia y en algunos casos, hiperlactacidemia.
3. DESÓRDENES METABÓLICOSa) Trastornos de la oxidación de ácidos grasos
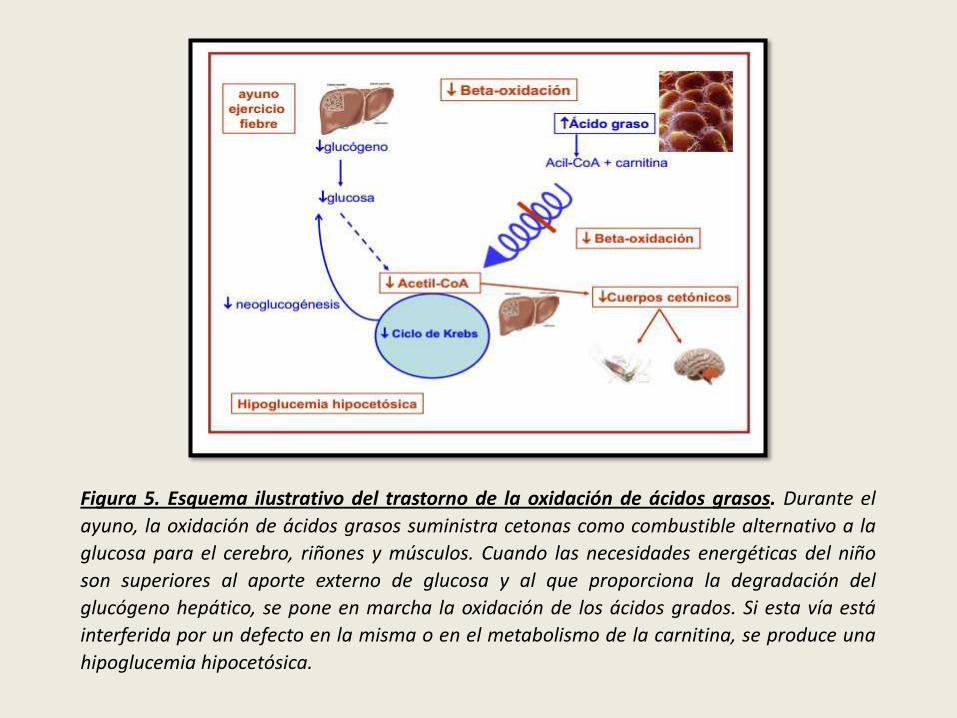
Figura 5. Esquema ilustrativo del trastorno de la oxidación de ácidos grasos. Durante el
ayuno, la oxidación de ácidos grasos suministra cetonas como combustible alternativo a la
glucosa para el cerebro, riñones y músculos. Cuando las necesidades energéticas del niño
son superiores al aporte externo de glucosa y al que proporciona la degradación del
glucógeno hepático, se pone en marcha la oxidación de los ácidos grados. Si esta vía está
interferida por un defecto en la misma o en el metabolismo de la carnitina, se produce una
hipoglucemia hipocetósica.

Aunque los niveles bajos de glucosa podrían explicar la encefalopatía que se
producen en estas deficiencias, se cree que ésta no es la única causa, ya que
algunos pacientes presentan encefalopatía en condiciones de normoglucemia.
Se cree que los efectos tóxicos de los ácidos grasos acumulados o de sus
metabolitos podrían contribuir a los efectos encefalopáticos. La alteración
muscular y hepática se podría justificar teniendo en cuenta que, tanto el
músculo como el hígado son los tejidos donde más eficazmente tiene lugar la β-
oxidación, y en consecuencia, los más afectados como consecuencia de una
interrupción de la misma.
3. DESÓRDENES METABÓLICOSa) Trastornos de la oxidación de ácidos grasos

3. DESÓRDENES METABÓLICOSb) Trastornos del almacenamiento del glucógeno
Las "enfermedades por almacenamiento de glucógeno" son un grupo de
trastornos hereditarios que se caracterizan por movilización deficiente del
glucógeno y depósito de formas anormales del mismo, conduciendo a
debilidad muscular e inclusive muerte.
Los trastornos de almacenamiento de glucógeno que afectan principalmente
el hígado, en general producen hepatomegalia e hipoglucemia, mientras que
los que afectan los músculos causan calambres y debilidad. Ambos tipos de
enfermedades pueden generar alteraciones renales y cardiovasculares.
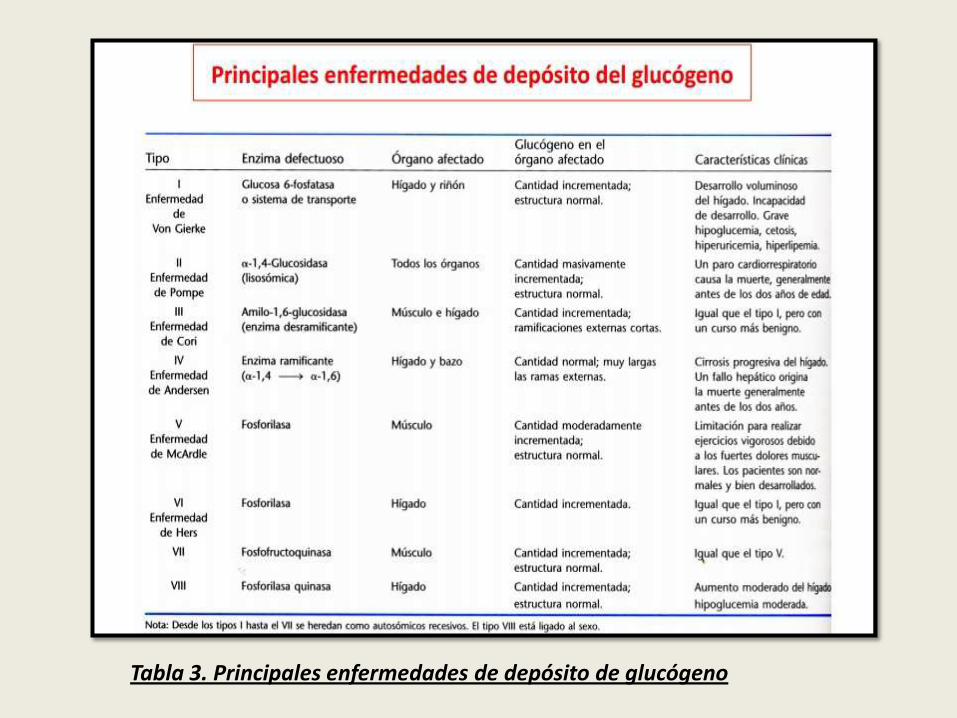
Tabla 3. Principales enfermedades de depósito de glucógeno

3. DESÓRDENES METABÓLICOSc) Trastornos de la gluconeogénesis
La gluconeogénesis es la ruta anabólica por la que tiene lugar la síntesis
de glucógeno, a partir del precursor más simple glucosa-6-fosfato. Se
lleva a cabo principalmente en hígado, y en menor medida, en el
músculo. Aparte de la deficiencia de la glucosa-6-fosfatasa (Enfermedad
de Von Gierke), se ha descrito defectos de otras enzimas que
interrumpen la vía de la gluconeogénesis: fructosa 1,6 bifosfatasa
(FBPasa), carboxiquinasa fosfoenolpiruvato (PEPCK) y la piruvato
carboxilasa.

La deficiencia de fructosa-1,6-bifosfatasa es un trastorno de herencia
autosómica recesiva que implica importantes alteraciones en la
gluconeogénesis.
La deficiencia de FBPasa se caracteriza por episodios repetidos de
hipoglucemia, hiperventilación, apnea, cetosis y acidosis metabólica, y
es, a menudo, letal en el período neonatal y la infancia. No
obstante, una vez efectuado el diagnóstico, la progresión de la
enfermedad es relativamente buena.
La excreción de glicerol en la orina puede ayudar a diferenciar la
deficiencia en FBPasa de otros errores metabólicos con síntomas
similares.
3. DESÓRDENES METABÓLICOSc) Trastornos de la gluconeogénesis

3. DESÓRDENES METABÓLICOSc) Trastornos de la gluconeogénesis
El déficit de fosfoenolpiruvato carboxiquinasa (PEPCK) es un trastorno
metabólico que se origina por un defecto en la gluconeogénesis. Es
una enfermedad poco frecuente. Los signos clínicos incluyen
hipotonía, hepatomegalia, problemas de desarrollo, acidosis láctica e
hipoglucemia. En la autopsia se evidencia la infiltración grasa tanto en
hígado como en riñones. Este trastorno se transmite de forma
autosómica recesiva.
El déficit de piruvato carboxilasa, pertenece al grupo de los defectos
del metabolismo intermediario de los hidratos de carbono asociados
con acidosis láctica.

3. DESÓRDENES METABÓLICOSc) Trastornos de la gluconeogénesis
La acidosis láctica se debe sospechar ante la presencia de la llamada
respiración de Kussmaul (respiraciones profundas y suspirosas), si no se corrige
puede dar lugar a deterioro de la conciencia progresivo hasta llegar al
coma, insuficiencia respiratoria, colapso cardiovascular, insuficiencia renal y
muerte.
Las manifestaciones clínicas pueden variar entre hipoglucemia en el
lactante, retraso psicomotriz que puede ser de intensidad variable, pero de
evolución progresiva y pronóstico mortal. Clínicamente se caracteriza por
vómitos, irritabilidad, crisis
convulsivas, letargia, hipotonía, ataxia, movimientos oculares
anormales, atrofia óptica y coma.
El diagnóstico se basa en determinar la actividad de la enzima en el hígado.
Está indicado tratamiento con tiamina para disminuir la acidosis láctica.

ACTITUD DIAGNÓSTICA DE LA HIPOGLUCEMIA NEONATAL
Anamnesis
Se requiere una anamnesis detallada de los factores de predisposición a la
hipoglucemia, incluyendo detalles del embarazo (diabetes
gestacional, restricción del crecimiento intrauterino), recepción neonatal
(sufrimiento fetal, asfixia al nacer), peso al nacer (bajo peso o macrosomía), la
edad gestacional (prematuro), incompatibilidad de grupo y factor
sanguíneo, medicamentos (Indometacina), mala absorción o condiciones de
desnutrición.
La relación entre el tiempo del episodio de hipoglucemia y la
alimentación, puede ser importante ya que puede dar una pista de la
enfermedad subyacente.
La cosanguineidad parental debe ser comprobada (trastornos
metabólicos, Hiperinsulinismo Congénito, etc., son más frecuentes en las
genealogías cosanguíneas), así como un historial familiar de muertes súbitas y
cualquier tipo de diabetes mellitus.

Examen físico
La macrosomía o Restricción de Crecimiento Intrauterino sugieren
Hiperinsulinismo Congénito como causa de la hipoglucemia.
Características dismórficas (como macroglosia, fístulas preauriculares, hemi-
hipertrofia) y organomegalia pueden sugerir Síndrome de Beckwith-
Wiedemann, que puede estar asociado a hiperinsulinismo.
La presencia de testículos no descendidos o micropene en un varón, la
hiperpigmentación, la ictericia y la los defectos de la línea media, pueden
indicar hipopituitarismo. Los genitales ambigüos podrían sugerir insuficiencia
suprarrenal.
La hiperventilación, hepatomegalia e ictericia son indicios de algunos
trastornos metabólicos.
ACTITUD DIAGNÓSTICA DE LA HIPOGLUCEMIA NEONATAL

ACTITUD DIAGNÓSTICA DE LA HIPOGLUCEMIA NEONATAL
Determinación del diagnóstico
La determinación precisa y rápida del estado glucémico es fundamental.
Dado que los signos clínicos de la hipoglucemia no son específicos, los bebés
que no se alimentan bien o que tienen síntomas neurológicos o respiratorios
o que están en situación de riesgo (Tabla 1), deben ser controlados.
Se les debe medir la glucemia antes de la alimentación, comenzando antes
de la segunda ingesta, y aproximadamente, cada 4 horas, hasta hallar por lo
menos, 2 mediciones satisfactorias y condiciones de estabilidad glucémica.
La hipoglucemia transitoria en las primeras horas después del
nacimiento, es relativamente común, pero si persiste y no responde a la
alimentación, se debe estudiar más profundamente.

Determinación del diagnóstico
Es fundamental, tomar las muestras apropiadas de metabolitos intermedios
y hormonas, en el momento de un episodio hipoglucémico. Dependiendo de
estos resultados, se podrán evaluar otros estudios.
La siguiente figura (Figura 6) muestra un Diagrama de Flujo para interpretar
los resultados clínicos y bioquímicos de neonatos con hipoglucemia.
ACTITUD DIAGNÓSTICA DE LA HIPOGLUCEMIA NEONATAL

Figura 6. Algoritmo para la interpretación de los resultados clínicos y bioquímicos de neonatos con hipoglucemia

TRATAMIENTO DE LA HIPOGLUCEMIA NEONATAL
La hipoglucemia es considerada una emergencia médica.
Si el recién nacido se encuentra asintomático y es capaz de tolerar laalimentación por vía oral o por sonda nasogástrica, se puede incrementar elvolumen y/o frecuencia de los alimentos, siempre y cuando la hipoglucemiano sea grave.
No se recomiendan soluciones orales de Dextrosa para este propósito, ya queno muestran ningún beneficio sobre la leche en el incremento de la glucemia.
Si el recién nacido permanece hipoglucémico o la hipoglucemia es grave, se lepuede administrar lentamente un bolo intravenoso de 10% de dextrosa (2-5ml/kg), seguida siempre de una infusión.

Mientras que se lleven a cabo los estudios para determinar la etiologíasubyacente, la glucosa en sangre debe mantenerse por arriba de 65mg/dl. Es posible que sea necesario aumentar la concentración deDextrosa y considerar un acceso central.
En un neonato, se considera normal un requerimiento entre 4 y 6mg/kg/min, que es el equivalente a la tasa de producción de glucosahepática normal. Cuando el requerimiento de Dextrosa supera los 8mg/kg/min, esto sugiere Hiperinsulinismo Congénito.
Estos niños deben ser trasladados a un Centro Especializado con loscuidados adecuados para evitar los episodios de hipoglucemia.
TRATAMIENTO DE LA HIPOGLUCEMIA NEONATAL

Si se sospecha de hipopituitarismo, el niño debe ser referido a unendocrinólogo pediátrico para evaluar la función pituitaria y recibir terapia desustitución.En los casos de niños con trastornos metabólicos, deben ser tratados enCentros con experiencia.
En conclusión, la mayoría de los casos de hipoglucemia neonatal sontransitorios, y suelen producirse en recién nacidos de alto riesgo.
Si la hipoglucemia es grave, persistente y/o recurrente, se requieren realizarestudios para determinar la condición subyacente. Las muestras de sangretomadas durante un episodio de hipoglucemia, son esenciales paraestablecer el diagnóstico y comenzar con el tratamiento adecuado.

DIAGNÓSTICO Y TRATAMIENTO DEL HIPERINSULINISMO
Tabla 4: Criterios para el diagnóstico de Hiperinsulinismo Congénito

DIAGNÓSTICO Y TRATAMIENTO DEL HIPERINSULINISMO CONGÉNITO
Diagnóstico
Para los niños que presentan hipoglucemia, el diagnóstico y tratamientoadecuado es esencial para evitar las secuelas neurológicas. Los indiciosclínicos para el diagnóstico incluyen características como recién nacidogrande para la edad gestacional o peso elevado, e hipoglucemia severay persistente que requiere flujos elevados de glucosa ( 1˃0 mg/kg/min).
Sin embargo, el fenotipo clínico del HIC también puede darse en bebéscon peso normal y menor requerimiento de glucosa.
El diagnóstico de HIC se realiza en base a la obtención de una muestra“crítica” de sangre obtenida durante un episodio espontáneo oprovocado de hipoglucemia.

Para disminuir la probabilidad de resultados falsos positivos, se haestablecido por convención, obtener la muestra de sangre con unaglucemia inferior a 50 mg/dl.
Durante la toma de muestra es importante un control estricto de laglucemia, signos vitales y estado neurológico para garantizar laseguridad del paciente. Al término de la prueba diagnóstico, laglucemia debe ser controlada cada 10-15 minutos, hasta asegurar queel paciente esté estable con una glucemia superior a 70 mg/dl.

Los niveles de insulina en plasma están aumentados de manerainadecuada durante la hipoglucemia; sin embargo, un error común en eldiagnóstico de HIC es que la concentración de insulina no siempre eselevada al momento de la hipoglucemia, por lo que el diagnóstico debebasarse en otros indicadores de la acción excesiva de la insulina. El hechode que los niveles de insulina no se encuentren aumentados al momentode la hipoglucemia, puede deberse a la liberación periódica deinsulina, que se pierde en una sola muestra, o a la rápida depuraciónhepática, de tal manera que el hígado es expuesto a altos niveles deinsulina y no se ve reflejado en la sangre venosa periférica. Esto tambiénpodría ser debido a la actividad de enzimas degradadoras de insulina quese encuentran presentes en muestras hemolizadas.
Resultados consistentes con la acción excesiva de insulina incluyensupresión en plasma de niveles de β-hidroxibutirato y de lasconcentraciones de ácidos grasos libres, así también como una respuestainadecuada de la glucemia al Glucagon ( 3˃0 mg/dl) en el momento de lahipoglucemia.
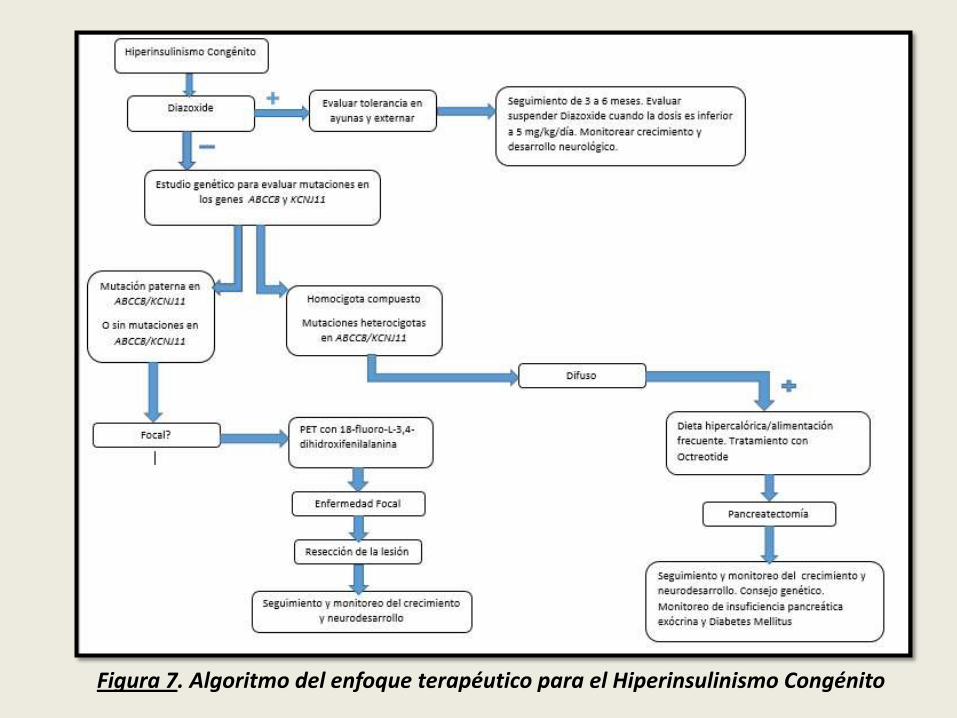
Figura 7. Algoritmo del enfoque terapéutico para el Hiperinsulinismo Congénito

DIAGNÓSTICO Y TRATAMIENTO DEL HIPERINSULINISMO CONGÉNITO
Tratamiento
El Diazoxide es la primera línea de tratamiento y la dosis es de 5-20
mg/kg/día, administrados por vía oral. Debido a la predisposición a la
sobrecarga de líquidos, la restricción de fluídos se combina con el diurético
Hidroclorotiazida (7-10 mg/kg/día). El Diazoxide inhibe la secreción de insulina
en las células β del páncreas, manteniendo los canales KATP abiertos.
En los pacientes que no responden al Diazoxide, la glucemia puede ser
estabilizada con Glucagón y/o Octreotide, y concentraciones elevadas de
glucosa. El Glucagón actúa mediante la liberación de las reservas de glucógeno
hepático y se puede administrar en bolo como infusión continua (5-10
mcg/kg/h) con el objetivo de estabilizar la glucemia.

El Octreotide es un análogo de la Somatostatina que activa los canalesKATP en las células β del páncreas, inhibiendo por ende, la secreción deinsulina. La dosis sugerida es de 5-20 mcg/kg/día, teniendo en cuentaciertas precauciones. La respuesta inicial al Octreotide es buena en lamayoría de los pacientes con Hiperinsulinsimo Congénito, pero luego deunas cuantas dosis, se puede producir taquifilaxia, dando lugar a unaterapia inadecuada a largo plazo.
También se ha demostrado que al reducir el flujo sanguíneo esplácnicode modo dosis-dependiente, puede afectar todo el tractogastrointestinal, y provocar enterocolitis necrotizante en recién nacidos.También, su administración prolongada puede provocar hepatitisinducida. Por ello, las enzimas hepáticas deben ser monitoreadas enforma rutinaria durante su administración, y se debe interrumpircuando se observa hepatitis sostenida.

Se ha descripto que el Acetato de Lanreotide (Somatuline Autogel), puedeser utilizado como alternativa del tratamiento farmacológico. Es unanálogo de la Somatostatina de larga duración. Se administra unainyección al mes (30 mg), lo que permite mejorar la calidad de vida de lospacientes.
Los niños que no responden al Diazoxide, requieren estudio genético. Lospacientes con mutaciones heterocigotas u homocigotas compuestos en losgenes ABCC8 o KCNJ11, presentan una forma difusa de enfermedad.
Las formas focales se heredan de manera esporádica, y se las harelacionado con la pérdida de la contribución del alelo materno del genABCC8 o del gen KCNJ11 de la región sometida a la impronta.

En las formas focales, el cromosoma materno está implicado en la expresión degenes que regulan el ciclo celular, y su pérdida produce la sobreexpresión degenes paternos que estimula la producción del factor de crecimiento IGF2, loque induce a una proliferación anormal de las células β afectadas en unahiperplasia adenomatosa focal. Estos pacientes son potencialmente curablescon resección de la lesión focal.
El estudio de imagen PET con 18-fluoro-L-3,4-dihidroxifenilalanina (18F-DOPA), permite confirmar y localizar con precisión la lesión focal para serresecada por vía laparoscópica, lo que resulta en la curación.
En la enfermedad difusa que no responde al tratamiento médico, se requeriráuna pancreatectomía casi total, pudiendo provocar a largo plazo diabetesmellitus e insuficiencia pancreática exócrina.

CENTROS ESPECIALIZADOS EN HIPERINSULINISMO CONGÉNITO
• The Children´s Hospital of Philadelphia Congenital HyperinsulinismCenter (E.E.U.U.) (En este Centro se realiza estudio de Pet Scan)
Dr. Charles Stanley ([email protected] )Dra. Diva De Leon- Crotchlow ([email protected] )
• Cook Children´s Hospital, Hyperinsulinism Center, Forh Worth, Texas(E.E.U.U)
Dr. Paul Thornton ( [email protected] )
• Great Ormond Street Hospital for Children Congenital HyperinsulinsmCenter (Gran Bretaña). En este Centro se realiza estudio de Pet Scan)
Dr. Khalid Hussain ( [email protected] )
• Charité Universidad de Medicina de Berlín (Alemania). En este Centro serealiza estudio de Pet Scan.
Dr. Oliver Blanckenstein ([email protected] )


















