
MÉTODO SCF DE HARTREE-FOCK -...
58
UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO FACULTAD DE QUÍMICA MÉTODO SCF DE HARTREE-FOCK Naytzé Ortiz Pastrana María del CarmenMorales Chamorro Miguel A. Olmedo Suarez Diana Alejandra Martínez Chávez
-
Upload
nguyentram -
Category
Documents
-
view
241 -
download
0
Transcript of MÉTODO SCF DE HARTREE-FOCK -...

UNIVERSIDAD NACIONAL
AUTÓNOMA DE MÉXICO
FACULTAD DE QUÍMICA
MÉTODO SCF DE
HARTREE-FOCK
Naytzé Ortiz Pastrana
María del CarmenMorales Chamorro
Miguel A. Olmedo Suarez
Diana Alejandra Martínez Chávez

La teoría de estructura electrónica abordada a través del método ab initio se enfoca a la predicción de las propiedades de los sistemas atómicos y moleculares.
Basada en las leyes de la mecánica cuántica
El tipo más común de cálculo ab initio es el método de Hartree Fock (HF), en el cual la aproximación principal es llamada aproximación de campo central.
INTRODUCCIÓN

Es un cálculo variacional, lo cual significa que las energías aproximadas calculadas son iguales o más grandes a la energía exacta.
El método de Hartree-Fock (HF) es empleado para resolver la ecuación de Schrödinger independiente del tiempo para átomos de múltiples electrones, moléculas y sólidos

Sirve para calcular de forma aproximada las funciones de onda y las energías de átomos e iones
Se basó en el modelo atómico de Bohr, donde la energía del estado atómico, para el átomo de Hidrógeno, depende del número cuántico principal , en unidades atómicas (u.a.) es dada como:

Se fundamenta en:
1. Asume la aproximación de Born-Oppenheimer (BO).
2. Los efectos relativistas son despreciados.
3. Asume que la solución variacional es combinación lineal de un número finito de funciones bases.

4. Asume que cada autofunción de energía se describe mediante un determinante de Slater.
5. La correlación electrónica es despreciada para electrones de espines opuestos y tomada en cuenta para electrones de espines paralelos.

El método de Hartree no respetaba el principio de antisimetría de la función de onda (usó el principio de exclusión de Pauli)
Al representar la función de onda total como un determinante de Slater, un determinante de orbitales de una partícula, satisface la propiedad de antisimetría de la solución exacta.
ANTECEDENTES

Por lo anterior, es apropiado para la aplicación del principio variacional.
La mecánica cuántica considera una serie de postulados con base en los cuales se puede obtener información del sistema en estudio.
A continuación se mencionan algunos postulados de importancia para el método.

Postulado 1
El estado de un sistema dinámico de N partículas esta descrito por una función ( q1,q2,…..q3N, t)
La cantidad *(q1 y q1 + dq) es proporcional a la probabilidad de encontrar a la partícula 1 entre q1 y q1 + dq1 ; a la partícula 2 entre q2 y q2 + dq2 y a la partícula N entre q3N y q3N + dq3N para un tiempo específico t.

Postulado 2
Para toda propiedad observable de un sistema, existe su correspondiente operador lineal Hermitiano y las propiedades físicas del sistema pueden ser inferidas a partir de las propiedades matemáticas asociadas a su operador.

Postulado 3
Sea A un operador correspondiente a un observable, entonces AΨs =asΨs, donde as es un número. Si un experimentador efectúa mediciones de la observable relacionada con el operador A, el resultado será siempre un valor propio de A. Sólo cuando la función de onda que describe al sistema coincide con la función propia de A, un experimento dará el mismo resultado en cada medición y éste coincidirá con el valor propio asociado a la función propia
HΨn=EnΨn Ecuación de onda del estado estacionario

Postulado 4
Si el sistema está descrito por una función que no es función propia del operador A, una serie de mediciones de la observable relacionada con el operador A no dará el mismo resultado. En lugar de eso se obtendrá una distribución de resultados donde el promedio estará dado por la expresión:
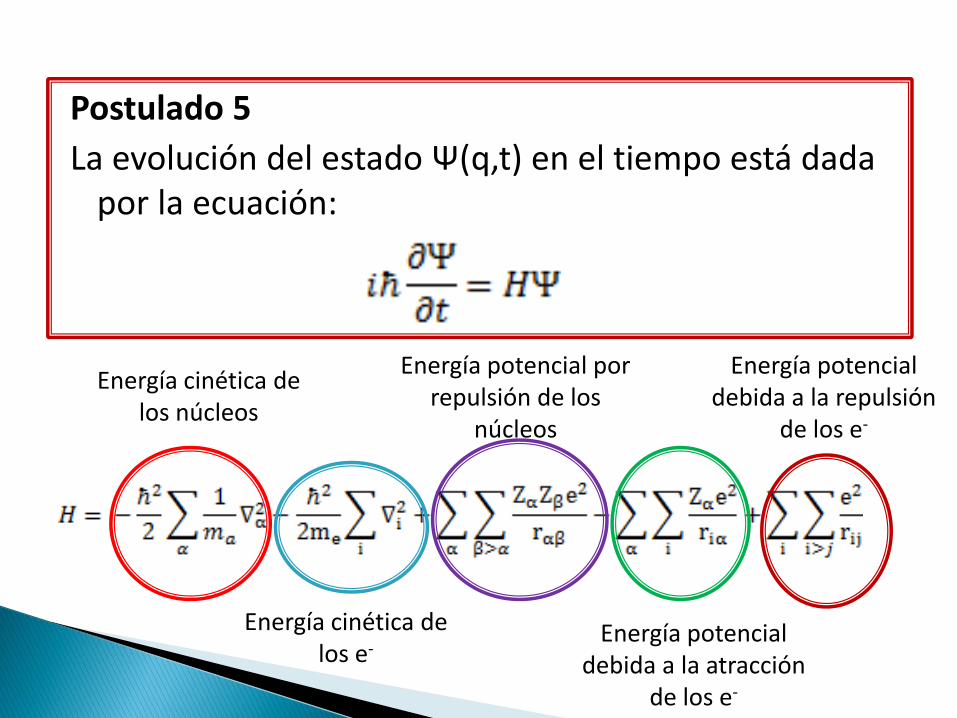
Postulado 5
La evolución del estado Ψ(q,t) en el tiempo está dada por la ecuación:
Energía cinética de
los núcleos
Energía cinética de los e-
Energía potencial por repulsión de los
núcleos
Energía potencial debida a la atracción
de los e-
Energía potencial debida a la repulsión
de los e-

El hamiltoniano de una molécula es muy complicado, pero la clave para efectuarla esta en las simplificaciones que se le pueden hacer.
La primera simplificación es la aproximación de Born-Oppenheimer y se basa en el hecho de que los núcleos son mucho más pesados que los electrones mα˃˃me
Aproximación de Born-Oppenheimer

El hamiltoniano se simplifica para obtener la ecuación de valores propios siguiente
𝐻𝑒𝑙 + 𝑉𝑁𝑁 Ψ𝑒𝑙 = 𝑈Ψ𝑒𝑙
Donde el hamiltoniano puramente electrónico 𝐻𝑒𝑙 tiene la forma:
𝐻𝑒𝑙 = −ħ2
2𝑚𝑒 𝛻𝑖
2
𝑖
− 𝑍𝛼 𝑒2
𝑟𝑖𝛼𝑖𝛼
+ 𝑒2
𝑟𝑖𝑗𝑖>𝑗𝑖

𝑉𝑁𝑁 = 𝑍𝛼 𝑍𝛽𝑒2
𝑟𝛼𝛽𝛽>𝛼𝛼
En la que 𝑉𝑁𝑁 es la repulsión entre los núcleos y 𝑟𝛼𝛽 es la
distancia entre los núcleos α y β. En esta aproximación las distancias 𝑟𝛼𝛽 no son variables sino constantes. La
energía U es la suma de la energía electrónica y la repulsión internuclear

Como las variables en la ecuación de Schrödinger son las coordenadas electrónicas y la cantidad 𝑉𝑁𝑁 es independiente de esas coordenadas, 𝑉𝑁𝑁es constante para una configuración nuclear dada, omitiendola:
𝐻𝑒𝑙Ψ𝑒𝑙 = 𝐸𝑒𝑙Ψ𝑒𝑙 𝑈 = 𝐸𝑒𝑙 + 𝑉𝑁𝑁

La ecuación de Schrödinger para el movimiento nuclear puede aproximarse por:
𝐻𝑁Ψ𝑁 = 𝐸Ψ𝑁
𝐻𝑁 = −ħ2
2
1
𝑚𝛼𝛻𝛼
2
𝛼
+ 𝑈
La energía E de la ecuación anterior es la energía total de la molécula y el hamiltoniano incluye los operadores tanto para la energía nuclear como la electrónica.

- Propiedad intrínseca de los electrones - Es el momento angular intrínseco caracterizado por el número cuántico S= ±1/2
19
Espin Electrónico (S)
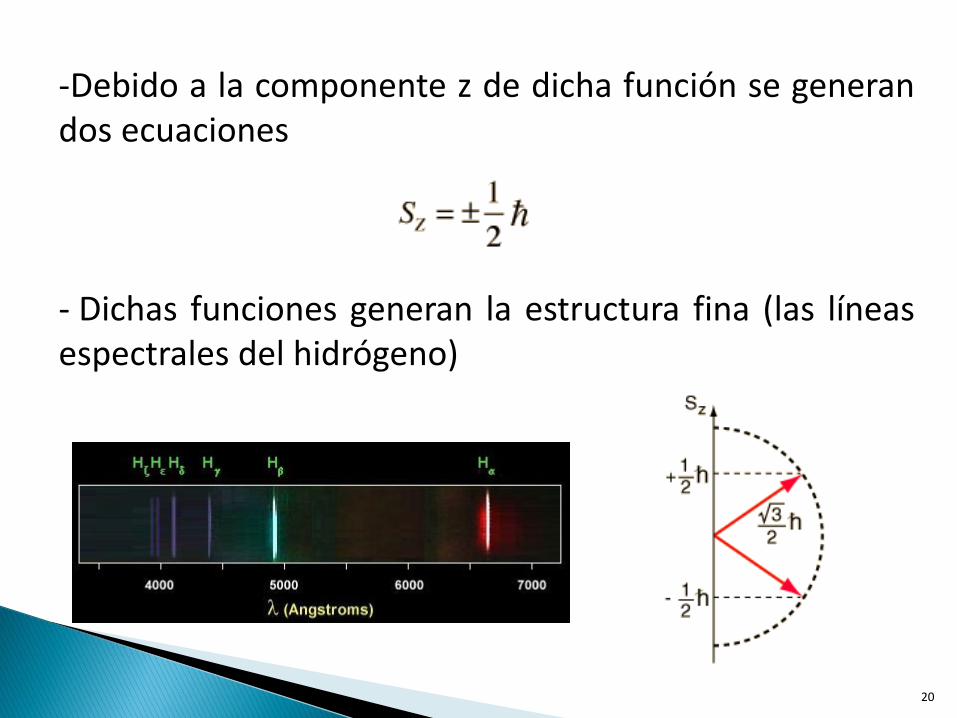
-Debido a la componente z de dicha función se generan dos ecuaciones
- Dichas funciones generan la estructura fina (las líneas espectrales del hidrógeno)
20

- Esto tiene como consecuencia una división en la energía del momento magnético
- Principio de exclusión de Pauli: establece que dos electrones no pueden tener todos sus números cuánticos iguales - Dicho principio es consecuencia directa del principio de antisimetría
21

-Alternativa para encontrar las soluciones de la ecuación de Schrodinger - Consta de la sustitución de un determinante de Slater en la ecuación de Schrödinger
F =E
- El hamiltoniano toma en cuenta la interacción coulombica entre un electrón y los que lo rodean. - El operador F emplea un promedio de interacciones
22
MÉTODO DE HARTREE-FOCK

La función de onda de Hartree-Fock - Escrita como el producto antisimétrico de espin-orbitales - Operador de Fock
- Consta de tres términos: Términos de energía cinética y potencial H, el operador coulómbico J y el operador de intercambio K
23

a) Energía cinética del electrón mas la energía potencial de la atracción entre el electrón 1 y los núcleos
b) Operador coulómbico Es el operador mono electrónico que define la repulsión generada por el electrón j f(1) Es el operador mono electrónico Es la función de onda mono electrónica del j-ésimo electrón
24

c) Operador de intercambio
Son las funciones de onda mono electrónicas sobre las que actúa el operador de intercambio, en función de la posición de los electrones uno y dos. Son las funciones de onda mono electrónicas del j-ésimo electrón, en función de la posición de los electrones uno y dos. Distancia entre los electrones 1 y 2
25
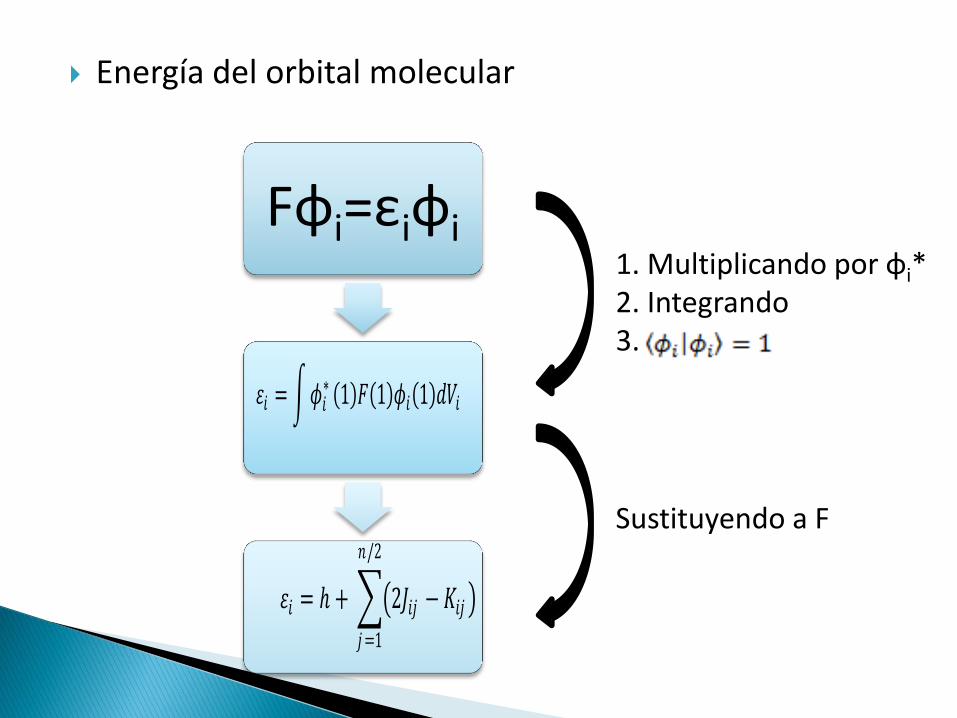
Energía del orbital molecular
Fφi=εiφi
𝜀𝑖 = 𝜙𝑖∗ 1 𝐹 1 𝜙𝑖 1 𝑑𝑉𝑖
1. Multiplicando por φi* 2. Integrando 3.
Sustituyendo a F
𝜀𝑖 = ℎ + 2𝐽𝑖𝑗 − 𝐾𝑖𝑗
𝑛/2
𝑗 =1
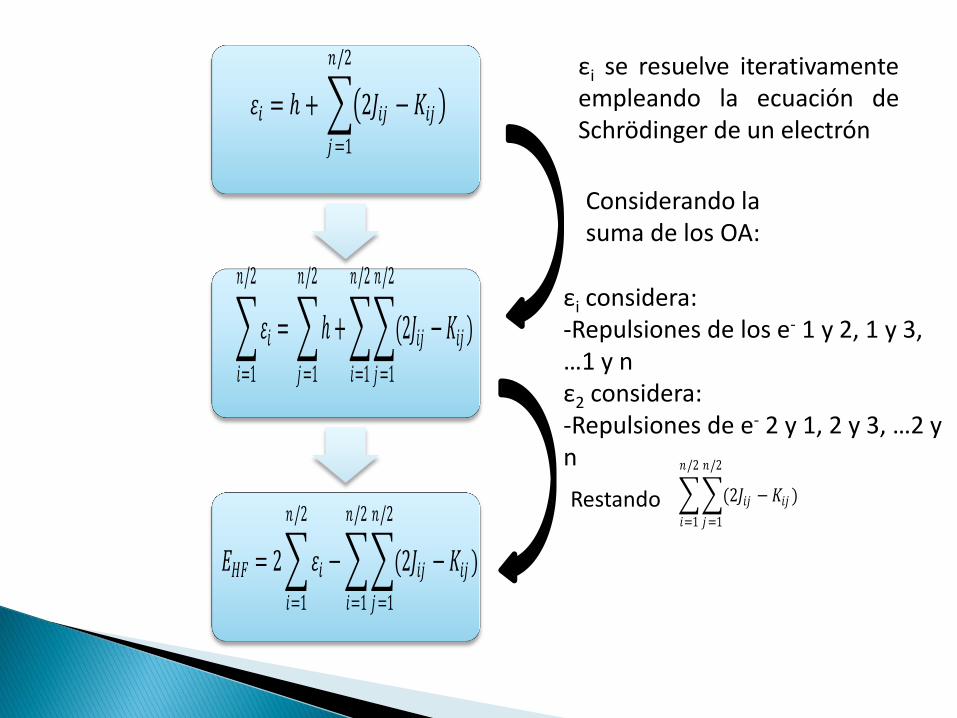
𝜀𝑖 = ℎ + 2𝐽𝑖𝑗 − 𝐾𝑖𝑗
𝑛/2
𝑗 =1
𝜀𝑖
𝑛/2
𝑖=1
= ℎ
𝑛/2
𝑗 =1
+ (2𝐽𝑖𝑗 − 𝐾𝑖𝑗 )
𝑛/2
𝑗 =1
𝑛/2
𝑖=1
Considerando la suma de los OA:
εi se resuelve iterativamente empleando la ecuación de Schrödinger de un electrón
εi considera: -Repulsiones de los e- 1 y 2, 1 y 3, …1 y n ε2 considera: -Repulsiones de e- 2 y 1, 2 y 3, …2 y n
(2𝐽𝑖𝑗 − 𝐾𝑖𝑗 )
𝑛/2
𝑗 =1
𝑛/2
𝑖=1
Restando
𝐸𝐻𝐹 = 2 𝜀𝑖 − (2𝐽𝑖𝑗 − 𝐾𝑖𝑗 )
𝑛/2
𝑗 =1
𝑛/2
𝑖=1
𝑛/2
𝑖=1

Coeficientes del orbital molecular
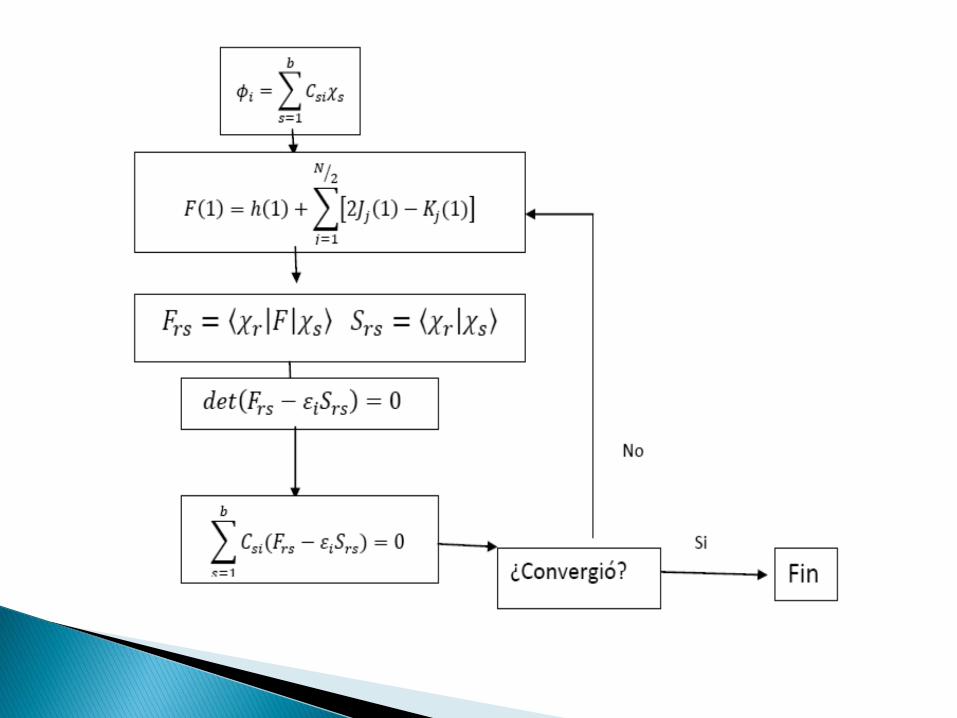
Para el cálculo se toma la propuesta de Roothan:
Sustituyendo en la Ec. de Hartree-Fock, se tiene:
Multiplicando por χ*r e integrando:
𝜙𝑖 = 𝐶𝑠𝑖𝜒𝑠
𝑏
𝑠=1
𝐶𝑠𝑖𝐹𝜒𝑠 =
𝑠
𝜀𝑖 𝐶𝑠𝑖𝜒𝑠
𝑎
.
𝐶𝑠𝑖
𝑏
𝑠=1
𝐹𝑟𝑠 − 𝜀𝑖𝑆𝑟𝑠 = 0
𝐹𝑟𝑠 = 𝜒𝑟 𝐹 𝜒𝑠 𝑆𝑟𝑠 = 𝜒𝑟 𝜒𝑠

Se forma un conjunto de b ecuaciones simultáneas con b incógnitas Csi
s= 1, 2, …,b describen orbitales moleculares φi
Para obtener una solución no trivial debe cumplirse:
𝑑𝑒𝑡 𝐹𝑟𝑠 − 𝜀𝑖𝑆𝑟𝑠 = 0
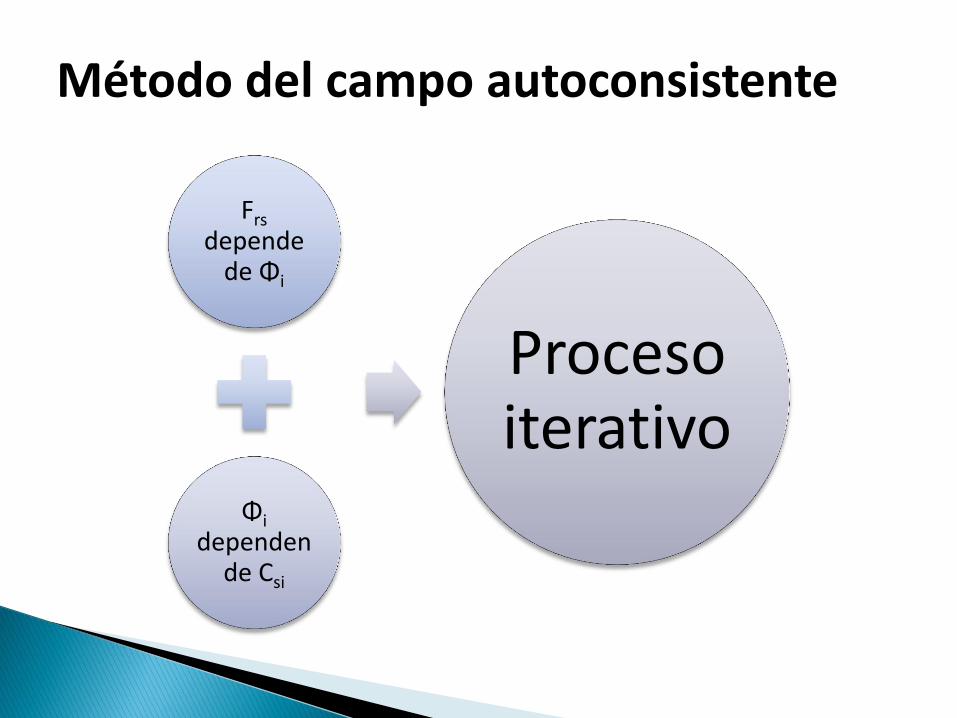
Frs depende
de Φi
Φi dependen
de Csi
Proceso iterativo
Método del campo autoconsistente

Comienza con un conjunto de orbitales iniciales que son utilizados para calcular el operador de Fock .
Se resuelve la ecuación secular y del resultado es posible obtener un nuevo conjunto de valores Csi
Se obtiene un nuevo conjunto de orbitales
Termina cuando la diferencia entre dos interacciones consecutivas es menor que el límite establecido en el parámetro de convergencia.
Método SCF

Primero se deben expresar los elementos de la matriz de Fock, 𝐹𝑟𝑠, en término de las funciones de base χ. De la definición del operador de Fock se tiene que:
𝐹𝑟𝑠 = 𝑋𝑟 1 F 1 𝑋𝑠 1 = 𝑋𝑟 1 h 1 𝑋𝑠 1 +
2 𝑋𝑟 1 𝐽𝑖 1 𝑋𝑠 1 − 𝑋𝑟 1 K𝑗 1 𝑋𝑠 1 𝑛/2𝑗=1
𝐽𝑗 1 𝜒 1 = 𝜒 1 𝜙𝑗 2
2
𝑟12 𝑑𝑣2
= 𝜒 1 𝐶𝑖𝑗∗ 𝐶𝑢𝑗
𝜒𝑟∗ 2 𝜒𝑢 2
𝑟12𝑢𝑖
𝑑𝑣2

Y multiplicando por 𝜒𝑟∗ e integrando sobre las
coordenadas del electrón 1 se obtiene:
𝑋𝑟 1 𝐽𝑖 1 𝑋𝑠 1 =
𝐶𝑖𝑗∗ 𝐶𝑢𝑗𝑢𝑖
𝜒𝑟∗ 1 𝜒𝑠 2 𝜒𝑖
∗ 2 𝜒𝑢 2
𝑟12𝑑𝑣1𝑑𝑣2 =
𝐶𝑡𝑗∗ 𝐶𝑢𝑗 rs tu 𝑏
𝑢=1𝑏𝑡=1
Que representa la integral rs tu de repulsión
Se puede obtener una expresión equivalente para el término de intercambio

𝑋𝑟 1 K𝑗 1 𝑋𝑠 1 = 𝐶𝑡𝑗∗ 𝐶𝑢𝑗 ru ts
𝑏
𝑢=1
𝑏
𝑡=1
Se pueden obtener los elementos de la matriz que forma al operador Frs en términos de las funciones de base,

𝐹𝑟𝑠 = ℎ𝑟𝑠 + 𝐶𝑡𝑗⋇ 𝐶𝑢𝑗 2 𝑟𝑠 𝑡𝑢 − 𝑟𝑢 𝑡𝑠
𝑛/2𝑗=1
𝑏𝑢=1
𝑏𝑡=1
𝐹𝑟𝑠 = ℎ𝑟𝑠 + 𝑃𝑡𝑢 𝑟𝑠 𝑡𝑢 −1
2𝑟𝑢 𝑡𝑠𝑏
𝑢=1𝑏𝑡=1
Donde
𝑃𝑡𝑢 = 2 𝐶𝑡𝑗⋇ 𝐶𝑢𝑗
𝑛/2𝑗=1
𝑃 = 𝜙𝑗⋇
𝑛
2𝑗=1
𝜙𝑗 =
2 𝐶𝑟𝑗⋇ 𝐶𝑠𝑗𝜒𝑟
⋇𝜒𝑠 = 𝑃𝑟𝑠𝜒𝑟⋇𝜒𝑠
𝑏𝑠=1
𝑏𝑟=1
𝑛
2𝑗=1
𝑏𝑠=1
𝑏𝑟=1
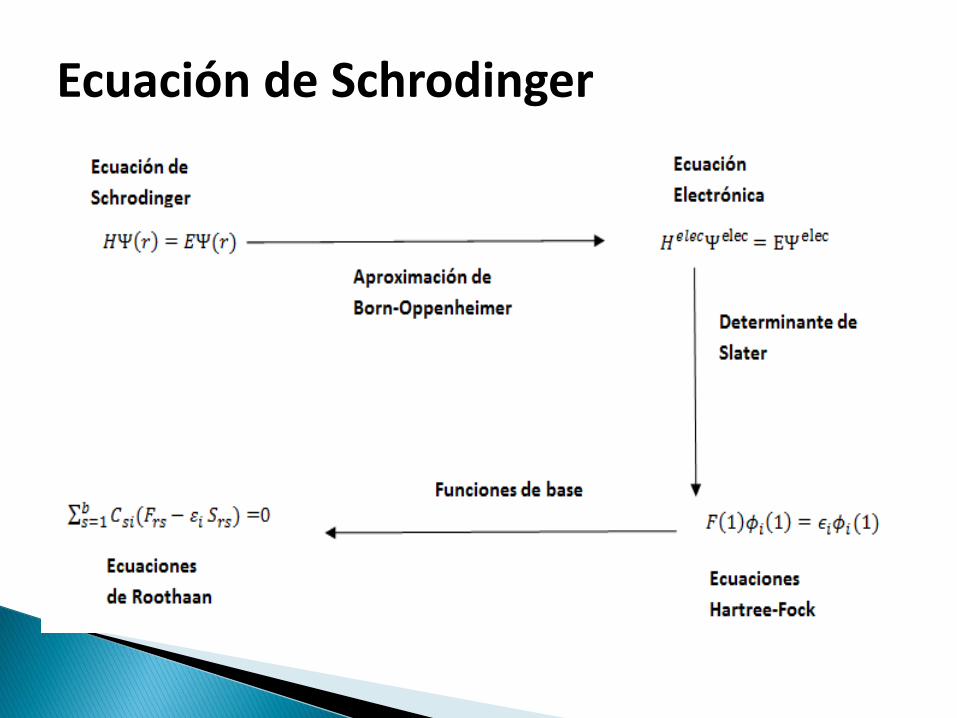
Ecuación de Schrodinger

Usando un conjunto de dos orbitales tipo Slater 1s con exponentes de orbitales ζ1= 1.45 y ζ2= 2.91
La funciones normalizadas del conjunto son (en unidades atómicas)
𝜒1 = 2𝜁13/2
𝑒−𝜁1𝑟𝑌00, 𝜒2 = 2𝜁1
3/2𝑒−𝜁2𝑟𝑌0
0, ζ1 = 1.45, ζ2 = 2.91,
EJEMPLO DESARROLLADO: Átomo de He

Se necesitan las integrales Frs y Srs. Las integrales de traslape Srsson
𝑆11 = 𝜒1 𝜒1 = 1, 𝑆22 = 𝜒2 𝜒2 = 1
𝑆12 = 𝑆21 = 𝜒1 𝜒2
= 4𝜁13/2
𝜁23/2
𝑒− 𝜁1+ 𝜁2 𝑟𝑟2𝑑𝑟∞
0
= 8𝜁1
3/2𝜁2
3/2
𝜁1 + 𝜁2 = 0.8366
Las integrales Frs dependen de 𝐻𝑟𝑠𝑐𝑜𝑟𝑒 , 𝑃𝑡𝑢 , y (rs|tu).

Empleando
𝐻𝑐𝑜𝑟𝑒 = −1
2𝛻2 −
2
𝑟= −
1
2𝛻2 −
𝜁
𝑟+ 𝜁 − 2 /𝑟 y
evaluando dichas integrales se encuentra que:
𝐻11𝑐𝑜𝑟𝑒 = 𝜒1 𝐻
𝑐𝑜𝑟𝑒 𝜒1 = −1
2𝜁1
2 − 2𝜁1 = −1.8488
𝐻22𝑐𝑜𝑟𝑒 =
1
2𝜁2
2 − 2𝜁2 = −1.5860

𝐻12𝑐𝑜𝑟𝑒 = 𝐻21
𝑐𝑜𝑟𝑒 = 𝜒1 𝐻𝑐𝑜𝑟𝑒 𝜒2
= −1
2𝜁2
22𝑆12 + 4 𝜁2 − 2 𝜁1
3/2𝜁2
3/2
𝜁1 + 𝜁2 2
=𝜁1
32𝜁2
32 4𝜁1𝜁2 − 8𝜁1 − 8𝜁2
𝜁1 + 𝜁2 3
= 1.8826

Muchas de las integrales de repulsión electrónica (rs|tu) son iguales entre sí. Para funciones de base reales, es posible mostrar que:
rs tu = sr tu = rs ut = sr ut = tu rs
= ut rs = tu sr = ut sr
Las integrales de repulsión electrónica son evaluadas usando la expansión 1/𝑟12 encontrando que:

11 11 = 5
8𝜁1 = 0.9062,
22 22 =5
8𝜁2 = 1.8188
11 22 = 22 11= 𝜁1
4𝜁1 + 4𝜁13𝜁2
2 + 𝜁1𝜁24 + 4𝜁1
2𝜁23 / 𝜁1 + 𝜁2
4
= 1.1826

12 12 = 21 12 = 12 21 = 21 21 =20𝜁1
3𝜁23
𝜁1+𝜁25 = 0.9536
11 12 = 11 21 = 12 11 = 21 11 =
16𝜁1
9/2𝜁2
3/2
3𝜁1+𝜁2 412𝜁1+8𝜁2
𝜁1+𝜁22
+ 9𝜁1+𝜁2
2𝜁12 = 0.9033
12 22 = 22 12 = 21 22 = 22 21
= 𝑙𝑎 𝑒𝑥𝑝𝑟𝑒𝑠𝑖𝑜𝑛 11 12 𝑐𝑜𝑛 1 𝑦 2 𝑖𝑛𝑡𝑒𝑟𝑐𝑎𝑚𝑏𝑖𝑎𝑑𝑎𝑠= 1.2980

El exponente optimo para un orbital atómico del helio
consiste de un orbital tipo slater 1s y es 27
16= 1.6875.
Como el exponente del orbital 𝜁1 es más cercano a 1.6875 que 𝜁2, es de esperarse que el coeficiente de χ1 en 𝜙1 = 𝐶11𝜒1 + 𝐶21𝜒2 sea sustancialmente más grande que el coeficiente de 𝜒2. Suponiendo ahora que 𝐶11
𝐶21≈ 2 la condición de normalización 𝜙1
2𝑑𝜏 = 1
da para coeficientes reales:

𝐶21 = 1 + 𝑘2 + 2𝑘𝑆12 −1/2
𝑑𝑜𝑛𝑑𝑒 𝑘 ≡ 𝐶11
𝐶21
La sustitución de k=2 y S12=0.8366 da 𝐶21 ≈ 0.3461 y 𝐶11 ≈ 2𝐶21 = 0.6922
Con n=2 y b=2, la ecuación da
𝑃11 = 2𝑐11
∗ 𝑐11, 𝑃12 = 2𝑐11
∗ 𝑐21 , 𝑃21 = 𝑃12,∗ 𝑃22 = 2𝑐21
∗ 𝑐21

La suposición inicial 𝑐11 ≈ 0.6922, 𝑐21 ≈ 0.3461 da los elementos de densidad inicial de matriz
𝑃11 ≈ 0.9583, 𝑃12 = 𝑃21 ≈ 0.4791, 𝑃22 ≈ 0.2396
𝐹11 = 𝐻11𝑐𝑜𝑟𝑒 +
1
2𝑃11 11 11 + 𝑃12 11 12
+ 𝑃22 11 12 −1
2 12 21

𝐹12 = 𝐹21 =
𝐻12𝑐𝑜𝑟𝑒 +
1
2𝑃11 12 11 +
𝑃123
212 12 −
1
2 11 22 +
1
2𝑃22 12 22
𝐹22 = 𝐻22𝑐𝑜𝑟𝑒 + 𝑃11 22 11 −
1
2 21 12 +
𝑃12 22 12 +1
2𝑃22 22 22
La sustitución de los valores de 𝐻𝑟𝑠𝑐𝑜𝑟𝑒 y las integrales
rs tu mencionadas previamente da:

𝐹11 = −1.8488 + 0.4531𝑃11 + 0.9033𝑃12 +0.7058𝑃22
𝐹12 = 𝐹21 = −1.8826 + 0.4516𝑃11 + 0.8391𝑃12 +0.6490𝑃22
𝐹22 = −1.5860 + 0.7058𝑃11 + 1.2980𝑃12 +0.9094𝑃22
La substitución de las suposiciones iniciales para los 𝑃𝑡𝑢´𝑠 da como los estimados iniciales de los 𝐹𝑟𝑠´𝑠

𝐹11 ≈ −0.813, 𝐹12 = 𝐹21 ≈ −0.892, 𝐹22 ≈ −0.070
El estimado inicial de la ecuación del determinante secular 𝐹𝑟𝑠−𝑆𝑟𝑠ℰ𝑖 = 0 es:
−0.813 − ℰ𝑖 −0.892 − 0.8366ℰ𝑖
−0.892 − 0.8366ℰ𝑖 −0.070 − ℰ𝑖≈ 0
0.3001ℰ𝑖2 − 0.609ℰ𝑖 − 0.739 ≈ 0
ℰ1 ≈ −0.854, ℰ2 ≈ 2.885

La sustitución de la menor raíz ℰ1 en la ecuación de Roothancon r=2 da:
𝑐11 𝐹21−ℰ1𝑆21 + 𝑐21 𝐹22−ℰ1𝑆22 ≈ 0
−0.177𝑐11 + 0.784𝑐21 ≈ 0 𝑐11/𝑐21 ≈ 4.42
De la sustitución de k=4.42 y 𝑆12 = 0.8366 en la condición de normalización se obtiene:
𝑐21 ≈ 0.189, 𝑐11 = 𝑘𝑐21 ≈ 0.836
Al emplear estos coeficientes mejorados se obtienen los elementos de densidad de matriz mejorados

𝑃11 ≈ 1.398, 𝑃12 = 𝑃21 ≈ 0.316, 𝑃22 ≈ 0.071
Y finalmente empleando estas 𝑃𝑡𝑢´𝑠 mejoradas da como los valores mejorados 𝐹𝑟𝑠
𝐹11 ≈ −0.880, 𝐹12 = 𝐹21 ≈ −0.940,𝐹22 ≈ −0.124
La ecuación secular mejorada es:
−0.880 − ℰ𝑖 −0.940 − 0.8366ℰ𝑖
−0.940 − 0.8366ℰ𝑖 −0.124 − ℰ𝑖≈ 0
ℰ1 ≈ −0.918, ℰ2 ≈ 2.810

Los valores mejorados de ℰ1 da los valores:
𝐶11
𝐶21≈ 4.61𝑐11 ≈ 0.842, 𝑐21 ≈ 0.183
Al realizar un nuevo ciclo de cálculos se obtienen los valores:
𝑃11 = 1.418, 𝑃12 = 𝑃21 = 0.308, 𝑃22 ≈ 0.067 𝐹11 = −0.881, 𝐹12 = 𝐹21 = −0.940,
𝐹22 = −0.124 ℰ1 = −0.918, ℰ2 = 2.809 𝑐11 ≈ 0.842, 𝑐21 ≈ 0.183

Estos últimos c´s son los mismos que los de los ciclos previos, así que el cálculo ha convergido y por tanto finalizado. El estado base del orbital atómico del He por SCF para este set base es:
𝜙1 = 0.842𝜒1 + 0.183𝜒2
Por último la energía de campo autoconsistente con n=2 y b=2 se escribe como

𝐸𝐻𝐹 = −0.918
+1
2 1.418 −1.8488
+ 2 0.308 −1.8826+ 0.067 −1.5860 + 0= −2.862 ℎ𝑎𝑟𝑡𝑟𝑒𝑒𝑠 = −77.9 𝑒𝑉

El método de Hartree-Fock, se basa en la aproximación orbital. Esta aproximación supone a la función de onda para un sistema de N electrones un producto antisimetrizado, de N orbitales monoelectrónicos.
Las ecuaciones de Hartree-Fock son ecuaciones de autovalores que determinan los orbitales moleculares, es decir son ecuaciones monoelectrónicas. Resolver la ecuación de Schrödinger dentro de la aproximación de HF equivale a resolver problemas monoelectrónicos.
La energía total no es igual a la suma de los autovalores de las ecuaciones de HF. Esto se debe al hecho que en la aproximación de HF las interacciones entre electrones se hallan consideradas por las integrales de Coulomb y de intercambio.
CONCLUSIONES

Para resolver las ecuaciones de HF los orbitales moleculares se ex-presan como combinación lineal de orbitales atómicos (método OM-CLOA) y el procedimiento se simplifica ya que es necesario obtener solamente los coeficientes de la combinación lineal.
El límite de base infinita, llamado límite de Hartree-Fock, no es igual al valor exacto de la energía, porque se ha utilizado la aproximación orbital.
La diferencia entre la energía exacta y la EHF, donde EHF es la energía límite de HF, se conoce como energía de correlación.
Metodologías post-HF

1. Cuevas, G. y Cortés, F. Introducción a la química computacional. Fondo de Cultura Económica, México, 2003. Páginas: 42-59.
2. Levine, I. Quantum Chemistry. Prentice-Hall. 4th Edition.
Páginas: 299-305 y 402-410. 3. Atkins and Friedman. Molecular Quantum Energy . 8th
Edition. Oxford University Press.2005. Páginas: 288-302.
4. Guía de trabajos prácticos.; Métodos de estructura electrónica. (2005) Tópicos de fisicoquímica de sistemas biológicos.
BIBLIOGRAFÍA


















