
PROYECTO FIN DE CARRERA
101
DESARROLLO PRÁCTICO INDUSTRIAL presentado por DIRECTORES: DR. JOSÉ LUIS VALVERDE PALOMINO D. DAVID SÁNCHEZ SÁNCHEZ-TOLEDO Ciudad Real, Junio 2010 ALBERTO MIRANDA DAHDAL UNIVERSIDAD DE CASTILLA-LA MANCHA FACULTAD DE CIENCIAS QUÍMICAS DEPARTAMENTO DE INGENIERÍA QUÍMICA M M M E E E J J J O O O R R R A A A D D D E E E L L L P P P R R R O O O C C C E E E S S S O O O D D D E E E R R R E E E F F F O O O R R R M M M A A A D D D O O O O O O X X X I I I D D D A A A T T T I I I V V V O O O D D D E E E E E E T T T A A A N N N O O O L L L P P P A A A R R R A A A L L L A A A P P P R R R O O O D D D U U U C C C C C C I I I Ó Ó Ó N N N D D D E E E H H H I I I D D D R R R Ó Ó Ó G G G E E E N N N O O O M M M E E E D D D I I I A A A N N N T T T E E E U U U N N N L L L E E E C C C H H H O O O A A A D D D I I I C C C I I I O O O N N N A A A L L L D D D E E E W W W A A A T T T E E E R R R G G G A A A S S S S S S H H H I I I F F F T T T
-
Upload
alberto-miranda -
Category
Documents
-
view
220 -
download
2
description
PROYECTO DE FIN DE CARRERA
Transcript of PROYECTO FIN DE CARRERA

DESARROLLO PRÁCTICO INDUSTRIAL
presentado por
DIRECTORES:
DR. JOSÉ LUIS VALVERDE PALOMINO
D. DAVID SÁNCHEZ SÁNCHEZ-TOLEDO
Ciudad Real, Junio 2010
ALBERTO MIRANDA DAHDAL
UNIVERSIDAD DE CASTILLA-LA MANCHA FACULTAD DE CIENCIAS QUÍMICAS
DEPARTAMENTO DE INGENIERÍA QUÍMICA
MMMEEEJJJOOORRRAAA DDDEEELLL PPPRRROOOCCCEEESSSOOO DDDEEE RRREEEFFFOOORRRMMMAAADDDOOO OOOXXXIIIDDDAAATTTIIIVVVOOO DDDEEE
EEETTTAAANNNOOOLLL PPPAAARRRAAA LLLAAA PPPRRROOODDDUUUCCCCCCIIIÓÓÓNNN DDDEEE HHHIIIDDDRRRÓÓÓGGGEEENNNOOO MMMEEEDDDIIIAAANNNTTTEEE
UUUNNN LLLEEECCCHHHOOO AAADDDIIICCCIIIOOONNNAAALLL DDDEEE WWWAAATTTEEERRR GGGAAASSS SSSHHHIIIFFFTTT

El presente Desarrollo Práctico Industrial ha sido realizado en
el Laboratorio de Catálisis del Departamento de Ingeniería Química de la
Universidad de Castilla-La Mancha, bajo la dirección del profesor J.L.
Valverde y David Sánchez.
Ciudad Real, España. Junio 2010

Agradecimientos
“A mis padres
Guido y Ana María.”

INDICE
1 RESUMEN ............................................................................................................... 1
2 INTRODUCCION ................................................................................................... 5
2.1 COMBUSTIBLES FÓSILES...................................................................................... 6
2.2 HIDRÓGENO ...................................................................................................... 12
2.2.1 Historia......................................................................................................... 12
2.2.2 Importancia del hidrógeno ........................................................................... 13
2.2.3 Métodos para la producción de hidrógeno .................................................. 15
2.2.3.1 Oxidación parcial de hidrocarburos ...................................................... 17
2.2.3.2 Reformado con vapor de agua .............................................................. 18
2.2.3.3 Reformado oxidativo con vapor de agua .............................................. 19
2.2.3.4 Gasificación de biomasa con posterior conversión ............................... 20
2.2.3.5 Electrólisis del agua. ............................................................................. 21
2.2.3.6 Energía nuclear ..................................................................................... 24
2.2.4 Almacenamiento del hidrógeno .................................................................... 26
2.2.5 Aplicaciones del hidrógeno .......................................................................... 29
2.3 BIOETANOL ....................................................................................................... 34
2.3.1 Importancia actual y perspectiva ................................................................. 34
2.4 REACCIÓN WATER GAS SHIFT .......................................................................... 35

2.4.1 Aplicaciones de la reacción Water Gas Shift ............................................... 37
3 OBJETIVOS Y ALCANCE DEL PROYECTO ................................................ 38
4 METODOLOGÍA EXPERIMENTAL ................................................................ 41
4.1 PREPARACIÓN DE LOS CATALIZADORES ............................................................ 42
4.1.1 Síntesis del soporte ....................................................................................... 42
4.1.2 Impregnación ............................................................................................... 43
4.1.3 Calcinación .................................................................................................. 43
4.2 TÉCNICAS DE CARACTERIZACIÓN ...................................................................... 44
4.2.1 Espectroscopía de emisión atómica de plasma acoplado por inducción
(ICP-AES)................................................................................................................ 44
4.2.2 Difracción de rayos X (DRX) ....................................................................... 45
4.2.3 Reducción térmica programada con hidrógeno (TPR) ................................ 47
4.2.4 Adsorción-desorción de nitrógeno a 77 K ................................................... 49
4.3 INSTALACIÓN EXPERIMENTAL ........................................................................... 50
4.3.1 Sección de alimentación ............................................................................... 52
4.3.2 Sección de reacción ...................................................................................... 52
4.3.3 Sección de análisis de productos ................................................................. 53
4.3.4 Parámetros de reacción ............................................................................... 54
4.4 PRODUCTOS QUÍMICOS UTILIZADOS .................................................................. 55
4.5 PROCEDIMIENTO EXPERIMENTAL ...................................................................... 56
5 RESULTADOS Y DISCUSIÓN ........................................................................... 58

5.1 CARACTERIZACIÓN DEL SOPORTE YSZ ............................................................. 59
5.1.1 Difracción de rayos X (DRX) ....................................................................... 59
5.1.2 Adsorción-desorción de N2 .......................................................................... 60
5.2 CARACTERIZACIÓN DE CATALIZADORES ........................................................... 61
5.2.1 Espectroscopía de emisión atómica de plasma acoplado por inducción
(ICP-AES)................................................................................................................ 61
5.2.2 Difracción de rayos X (DRX) ....................................................................... 61
5.2.3 Adsorción-desorción de N2........................................................................... 64
5.2.4 Reducción térmica programada con hidrógeno (TPR) ................................ 67
5.3 ENSAYOS CATALÍTICOS ..................................................................................... 69
5.3.1 Efecto de la temperatura .............................................................................. 69
5.3.2 Efecto del tamaño de partícula .................................................................... 71
5.3.3 Estudio de doble lecho ................................................................................. 72
5.3.3.1 Efecto de la velocidad espacial ............................................................. 75
6 CONCLUSIONES ................................................................................................. 78
7 RECOMENDACIONES ....................................................................................... 81
8 ANEXO .................................................................................................................. 84
8.1 ÍNDICE DE FIGURAS .................................................................................... 85
8.2 ÍNDICE DE TABLAS ..................................................................................... 87
9 BIBLIOGRAFIA ................................................................................................... 89

1 RESUMEN

1. RESUMEN
2
El presente trabajo está enmarcado en una línea de investigación llevada a cabo
actualmente en el Departamento de Ingeniería Química de la Facultad de Ciencias
Químicas de la Universidad de Castilla-La Mancha.
Concretamente, esta investigación es fruto de un proyecto financiado por la
Junta de Comunidades de Castilla-La Mancha y se centra en la producción de hidrógeno
por reformado oxidativo de etanol con vapor de agua, con catalizadores de níquel
soportados sobre YSZ. Así mismo, este proceso se mejoró con la adición de un segundo
lecho, formado por un catalizador comercial de CrO3 y Fe2O3 en el cual tiene lugar la
reacción Water Gas Shift (WGS). La finalidad del doble lecho es aprovechar el CO
producido en el primer lecho, para convertirlo en H2 y aumentar el rendimiento global
del proceso.
El reformado oxidativo de etanol con vapor de agua es un proceso complejo en
el que intervienen un gran número de reacciones, por lo que es frecuente la aparición de
productos indeseables. Dado que unas reacciones son favorecidas sobre otras
dependiendo del catalizador utilizado y de las condiciones de reacción, se planteó como

1. RESUMEN
3
principales objetivos de la investigación concluir qué catalizador era el más adecuado,
en qué condiciones y cómo podía aumentarse el rendimiento global del proceso con la
adición de un segundo lecho.
A continuación se resume el esquema planteado para el desarrollo de la presente
investigación:
1. Síntesis de catalizadores.
2. Caracterización de los catalizadores.
3. Ensayos catalíticos
a. Un lecho. Estudio del efecto de la temperatura y del tamaño de partícula
de la fase activa de níquel.
b. Doble lecho. Estudio del rendimiento global del proceso y del efecto de
la velocidad espacial.
4. Discusión de resultados y conclusiones.
La fase metálica de níquel se incorporó sobre el soporte mediante el método de
impregnación a humedad incipiente, utilizando disoluciones acuosas de Ni(NO3)2 · 6
H2O como precursor. Posteriormente se procedió a su calcinación para eliminar los
grupos nitrato.
Todos los catalizadores se caracterizaron utilizando diversas técnicas de análisis:
Análisis de difracción de rayos X (DRX).
Análisis de espectroscopía de emisión atómica de plasma acoplado por
inducción (ICP-AES).
Análisis de reducción térmica programada (TPR).
Adsorción-desorción de N2 a 77 K.
Se llevó a cabo la preparación de los catalizadores con distintas cantidad de fase
activa (10%, 15% y 23%). El tamaño de partícula de níquel depende de manera directa
de la cantidad impregnada en el catalizador, obteniéndose mayores diámetros a medida
que aumenta dicha cantidad. Se estudió el efecto del tamaño de partículas sobre la
distribución de los productos de reacción.

1. RESUMEN
4
Se ordenaron todos los resultados obtenidos y se llevaron a cabo los cálculos
pertinentes para la obtención de los principales parámetros de reacción (conversión,
selectividad, relación CO2/CO y relación H2/CO). El valor de estos parámetros
dependerá en gran medida de las propiedades de cada catalizador, de la influencia de la
estructura porosa del soporte y de las condiciones de reacción.
Los resultados obtenidos más relevantes fueron los siguientes:
1. La adición de mayor carga metálica al catalizador Ni-YSZ suponía una
impregnación de partículas de níquel de mayor tamaño.
2. Para un mismo soporte, la temperatura de reducción de los catalizadores era
menor para aquel con mayor carga metálica. Al haber más cantidad de Ni, la
dispersión era menor y, por lo tanto, las especies metálicas eran más fáciles de
reducir al disminuir la interacción entre el soporte y éstas.
3. Para todos los catalizadores sintetizados, tras la incorporación de la carga
metálica, el volumen de poro y el diámetro de poro aumentaba, debido a la
acidez de la disolución de impregnación del metal, que atacaba la estructura del
soporte haciendo que los valores de las propiedades texturales aumentasen.
4. El proceso de reformado oxidativo de etanol ofreció conversiones muy elevadas
en todos los casos estudiados de temperaturas y tamaños de las partículas de
níquel, superándose siempre el 99%.
5. A partir de 400ºC, los resultados obtenidos de selectividad de H2 eran similares.
Una disminución de la temperatura aumentaba la relación CO2/CO.
6. A medida que disminuye el tamaño de partícula de la fase activa de níquel
aumenta la selectividad de hidrógeno y la relación CO2/CO. Los motivos
principales de la tendencia apreciada están actualmente en estudio y no se
encontraron referencias bibliográficas con qué compararlo.
7. La adición de un segundo lecho con catalizador WGS aumentó el rendimiento
global del proceso hacia la producción de hidrógeno. La relación CO2/CO se
incrementó considerablemente debido a la conversión del monóxido de carbono.
8. A medida que disminuía la velocidad espacial del catalizador WGS, y por tanto
aumentaba el número de centros activos, se incrementaba la relación H2/CO
debido al desplazamiento del equilibrio hacia la conversión de monóxido de
carbono para producir hidrógeno.

2 INTRODUCCION

2. INTRODUCCIÓN
6
2.1 Combustibles fósiles
A lo largo de la historia de la humanidad, las diversas civilizaciones han
empleado la energía de diferente manera; calentamos nuestras casas y oficinas con
combustibles fósiles, mantenemos nuestras fábricas y nuestros sistemas de transporte
con combustibles fósiles, iluminamos nuestras ciudades y nos comunicamos a distancia
con electricidad generada a partir de combustibles fósiles, construimos nuestros
edificios con materiales hechos con combustibles fósiles, tratamos nuestras
enfermedades con medicamentos derivados de combustibles fósiles, almacenamos
nuestros excedentes en contenedores de plástico y embalajes hechos de combustibles
fósiles y manufacturamos nuestras ropas y aparatos domésticos con la ayuda de
productos petroquímicos. Prácticamente todos los aspectos de nuestra vida moderna
extraen su energía de los combustibles fósiles, derivan materialmente de ellos o reciben
su influencia de algún otro modo.
Se sabe que los recursos fósiles son finitos, su extinción puede ser más próxima
de lo que imaginamos y al parecer no hemos tomado suficiente conciencia de que esto
pueda ser así. Los cálculos más optimistas hablan de un horizonte de 42 añosi para que
los recursos "toquen fondo"; y los menos optimistas, entre 8 y 18 años; quedando la

2. INTRODUCCIÓN
7
mayor parte de las reservas en Oriente Medio, por lo tanto, es solo una cuestión de
tiempo que el mundo pase a depender del golfo Pérsico para satisfacer sus crecientes
necesidades de petróleo. Durante los próximos años, el descenso de la producción
petrolera de los yacimientos de Rusia, Mar del Norte, Alaska y África Occidental, que
hoy satisfacen las economías de EEUU y de Europa (sólo EEUU, con el 5% de la
población mundial, consume el 26% del petróleo de todo el mundo), agotarán los
recursos inevitablemente. Hay más de 40.000 yacimientos petrolíferos conocidos en el
mundo, pero solo 40 de ellos se consideran gigantes; es decir, con más de 5.000
millones de barriles de petróleo; de éstos, 26 están en el golfo Pérsico y se hallan aún en
fase ascendente de producción, a diferencia de los de EEUU y Rusia, en donde han
tocado techo o están en fase descendente.
Las sociedades, como lo hacen los seres vivos en la naturaleza, luchan por
remontarse a la segunda ley de la termodinámica, o a la degradación ineluctable. Esta
curva de entropía se altera al introducir nueva energía para sostener el orden energético
y por consiguiente el orden social. En este sentido, cabe entonces la preocupación de
¿cuánta energía requieren las sociedades para conservar su estilo de vida actual, frente
al hecho de que los recursos energéticos se están agotando? Si la energía no se crea ni se
destruye, sólo se transforma, el problema radica en el sentido de esta transformación, el
cual va de una energía disponible a una no-disponible. Algunos científicos advierten
que... "Las sociedades que más duran son aquellas que consiguen el mejor equilibrio
entre el balance de la naturaleza y de la sociedad humana, dentro de los límites que
impone inevitablemente la segunda ley"ii; de lo contrario, las sociedades colapsan.
Tres problemas se configuran frente a esta situación. El primero radica en que
una mayor concentración y centralización de poder en un número menor de
instituciones puede resultar poco flexible a la hora de enfrentarse a nuevos retos; en
segundo lugar, el auge del fundamentalismo islámico en Oriente Medio, preocupa
respecto de las decisiones sobre las últimas reservas del petróleo; y, finalmente, el
problema del calentamiento global del planeta, por la quema de combustibles fósiles.
Estos tres aspectos se configuran como cruciales para determinar las perspectivas de la
civilización humana en el siglo XXI. De la forma como se afronte este "punto de
inflexión" en la curva del régimen energético, dependerá que tengamos un renacimiento
como civilización, o un deterioro progresivo de la misma.

2. INTRODUCCIÓN
8
Una de las nuevas apuestas energéticas de muchos científicos e instituciones en
el mundo, es la del uso del hidrógeno como fuente energética. El hidrógeno es un
combustible eterno que no contamina y aunque se halla prácticamente en todas partes,
raramente aparece en la naturaleza en estado libre, por lo que debe ser extraído. Las
diversas formas de producirlo, que aunque pueden involucrar el uso de energías
procedentes de hidrocarburos, se inclinan hacia la utilización de energías renovables,
como la fotovoltaica, la eólica, la hidráulica y la geotérmica.
Como se acaba de exponer, más del 80 % de la demanda energética mundial se
abastece con combustibles fósiles. Sin embargo, la utilización de este tipo de
combustibles lleva asociada una serie de inconvenientes:
• El desajuste entre la localización de la demanda y de la producción.
En 2007, la Unión Europea consumió 1745 Mtep de los distintos tipos de
energías, mientras que sólo produjo 890 Mtep. El grado de autoabastecimiento
energético disminuyó desde el 57 % en 1990 hasta 51 % en 2007, debido a la creciente
importación de todas las fuentes energéticas primarias, especialmente gas y el carbón,
así como el 77% del petróleoiii
. De mantenerse la tendencia actual, las importaciones de
gas aumentarían un 80 % en los próximos 25 años y, si no se consigue otorgar una
mayor competitividad a la energía autóctona, en los próximos 20 o 30 años un 70 % de
las necesidades energéticas de la Unión Europea se satisfarán mediante productos
importadosiv
. Esto demuestra que la dependencia de Europa respecto de las
importaciones va en aumento y, si no se frena el aumento del consumo en los
principales sectores de expansión, que son el transporte, los hogares y los servicios, la
dependencia energética de la Unión seguirá aumentandov.
Concretamente en España, el sistema de energía se basa fundamentalmente en la
importación de carbón, gas natural y, sobre todo, petróleo. Sin embargo, las principales
fuentes de energía localmente disponibles son la nuclear y renovables, utilizadas para el
consumo localvi
. Además, el aumento de la demanda por encima de la producción
interior, ha hecho que el grado de autoabastecimiento energético se sitúe en el 18,7 %
en 2007.

2. INTRODUCCIÓN
9
• La localización de las reservas en zonas geográficas de elevada inestabilidad
política.
Las reservas de petróleo y gas natural están concentradas en número reducido de
países. Más del 70 % de las reservas mundiales de petróleo están localizadas en países
miembros de la Organización de Países Exportadores de Petróleo (OPEP). Según lo
previsto, en el año 2020, la OPEP cubrirá sólo el 50 % de las necesidades de la Unión
Europea, con una producción del orden de 55 millones de barriles por día, frente a los
32 millones de barriles por día del año 2000. Por otro lado, aproximadamente la mitad
del consumo de gas de la Unión Europea se satisface con gas procedente de sólo tres
países (Rusia, Noruega y Argelia). En términos geopolíticos, el 45 % de las
importaciones de petróleo en Europa proceden de Oriente Medio, y el 40 % de las
importaciones de gas natural, de Rusia, regiones situadas bajo la amenaza de la
inseguridadiv,v
. Por tanto, los aspectos geopolíticos no facilitan el buen funcionamiento
del mercado de los combustibles fósiles.
• Los problemas de contaminación ambiental asociados a su combustión.
Según el Grupo Intergubernamental de Expertos sobre el Cambio Climático
(IPCC)vii
, desde 1900 se acelera el calentamiento de la atmósfera y la serie de
temperaturas récord alcanzadas los últimos 25 años sería una prueba tangible de ello. La
Tierra se ha recalentado en un promedio de 0,3 a 0,6 °C, debido a la intensificación de
un fenómeno natural y esencial para la supervivencia en la Tierra: el efecto invernadero.
La dependencia de las energías fósiles se traduce en emisiones de gases de efecto
invernadero, destacando entre todas ellas el CO2
(80 % de contribución), producto de su
combustiónv. Puesto que la demanda global de energía sigue creciendo, se espera que
las emisiones globales de dióxido de carbono se incrementen un 1,7 % por añoii, de
modo que la utilización mundial de combustibles fósiles para las necesidades
energéticas darán lugar a problemas medioambientales críticos en los ecosistemas de
todas las regiones del mundoviii
. El IPCC estima que el aumento de temperaturas podrá
alcanzar entre 1,4 y 5,8 ºC al final del presente siglo si no se adopta medida alguna al
respectoiv
.

2. INTRODUCCIÓN
10
Un sistema energético basado en el uso de combustibles fósiles conduce a dos
problemas fundamentales: la dependencia energética y el deterioro del medioambiente.
Por eso, las políticas energéticas de los países desarrollados están orientadas a lograr los
objetivos básicos de seguridad en el abastecimiento energético, contribución de la
energía al aumento de la competitividad de la economía y la integración de los objetivos
medioambientalesvix
. Para ello, se requiere una normativa que apoye estos objetivos.
La regulación destinada a la protección del medio ambiente a nivel
internacional, en particular, las relativas a las emisiones de gases de efecto invernadero,
está teniendo una importancia creciente en las actividades energéticas. Esto está
llevando a la realización de importantes inversiones, al desarrollo de tecnologías más
limpias y al diseño de nuevas estrategias en el sectoriii
. Esta lucha contra el cambio
climático condujo a la adopción de unos objetivos provisionales en la Cumbre de la
Tierra de Río, celebrada en 1992 bajo el amparo de las Naciones Unidas. El Convenio
de Río fue seguido del Protocolo de Kioto de la Convención Marco de las Naciones
Unidas sobre el Cambio Climáticox, adoptado el 11 de diciembre de 1997 y con entrada
en vigor el 16 de febrero del 2005 (tras ratificarlo un número suficiente de países), por
el cual los países industrializados y de economías en transición se comprometieron de
forma más precisa y vinculante a limitar las emisiones de los seis gases de efecto
invernadero (CO2, CH4, N2O, HFCs, PFCs y SF6) entre 1990 y el período 2008-2012.
Entre los compromisos de reducción de emisiones más relevantes se puede citar:
Estados Unidos -7%, Japón -6%, Rusia 0%, Australia +8%, etc. La Unión Europea se
comprometió, en un primer momento, a estabilizar sus emisiones de CO2 en el año 2000
al nivel de las de 1990 y, después, a reducir globalmente sus emisiones de gases de
efecto invernadero hasta el período 2008-2012 en un 8 % con respecto al nivel de 1990,
lo que equivale a una reducción de 346 millones de toneladas de CO2. Dentro de la
Unión Europea se celebró un acuerdo de reparto de la carga en virtud del cual Alemania
debe aplicar una reducción del 21% y el Reino Unido del 12,5%, mientras que Francia y
Finlandia pueden limitarse a estabilizar sus emisiones y a España se le permitía un
incremento de hasta el 15%.
A nivel europeo, existen diversos documentos que proponen una estrategia
energética para Europa basada en el equilibrio entre desarrollo sostenible,
competitividad y seguridad de abastecimiento:

2. INTRODUCCIÓN
11
- Libro Verde: Estrategia europea para una energía sostenible, competitiva
y seguraiv
.
- Libro Verde: Hacia una estrategia europea de seguridad del
abastecimiento energéticov.
- Libro Blanco: La política europea de transportes de cara al 2010; la hora
de la verdadix
.
En ellos, se propone:
- Un mercado interior de la energía realmente competitivo, que garantice la
seguridad de suministro a precios más bajos. Esto requiere la solidaridad entre Estados
miembros, actuando de forma integrada, aunque cada estado tomaría sus decisiones en
función de sus propias preferencias nacionales.
- Las emisiones globales de gases de efecto invernadero deberían alcanzar
su punto culminante a más tardar en 2025 y, a continuación, reducirse entre un 15 y un
50 % respecto de los niveles de 1990. La envergadura de este reto exige que Europa
actúe de inmediato, especialmente en los ámbitos de la eficiencia energética y de las
fuentes de energía renovable. Además, las medidas que se adopten en estos dos sectores
contribuirán a reforzar la seguridad de abastecimiento y a limitar la creciente
dependencia de la energía importada.
- Asimismo, se especifica que las medidas políticas deben tener como
objetivo prioritario:
- La reducción del consumo, racionalizando para ello el uso del vehículo
particular clásico en los centros urbanos, y el fomento de los transportes urbanos
limpios, así como fomentando los esfuerzos para la utilización del hidrógeno como
carburante en los vehículos de mañana.
- El aumento de la cuota de las energías renovables y vectores energéticos
de baja emisión de carbono, en particular combustibles alternativos para el transporte,
ya que presentan un potencial nada desdeñable para reforzar la seguridad de
abastecimiento de Europa, aunque su desarrollo exige esfuerzos políticos y económicos
extremadamente importantes. A medio plazo, las energías renovables son la única
fuente de energía en que la Unión Europea dispone de cierto margen de maniobra para
aumentar la oferta en las circunstancias actuales.

2. INTRODUCCIÓN
12
Para posibilitar el cumplimiento del compromiso adquirido entre España y la
Unión Europea sobre el protocolo de Kioto, fue aprobado el Plan Nacional de
Asignación de Derechos de Emisión 2005-2007xi
para los sectores de la Directiva del
Comercio de Emisiones, cuyo objetivo básico es la reducción de aproximadamente el
0,2 % respecto a las emisiones de 2002, y se intensificará en un Plan Nacional de
Asignación 2008-2012, periodo durante el cual el promedio de las emisiones no debería
sobrepasar en más de un 24% las de 1990. Como medida adicional, en materia de
eficiencia energética, se aprobó el Plan de Acción 2005-2007 para la Estrategia de
Ahorro y Eficiencia Energética 2004-2012xii
, con el objetivo de conseguir ahorros
energéticos equivalentes al 8,5% del consumo y al 20% de las importaciones de petróleo
actuales. Además, se aprobó el Plan de Energías Renovables 2005-2010xiii
, que
constituye la revisión del Plan de Fomento de las Energías Renovables en España 2000-
2010 hasta entonces vigente. Con esta revisión, se trata de mantener el compromiso de
cubrir con fuentes renovables al menos el 12% del consumo total de energía en 2010,
así como de incorporar los otros dos objetivos indicativos (29,4% de generación
eléctrica con renovables y 5,75% de biocarburantes en transporte para ese año)
adoptados con posterioridad al anterior Plan.
2.2 Hidrógeno
2.2.1 Historia
El hidrógeno es el elemento más abundante y ligero de la naturaleza, si bien en
el planeta Tierra únicamente se encuentra formando moléculas con otros elementos
como el agua o los hidrocarburos. La producción de hidrógeno, pues, requiere contar
con materia prima que lo contenga y el desarrollo de tecnologías capaces de extraerlo.
La primera referencia histórica escrita acerca del hidrógeno procede de
Paracelso, célebre alquimista, quien en el s. XVI observó un aire que se desprendía al
hacer reaccionar un ácido sobre hierro, el cual era inflamable. Sin embargo no fue hasta
mucho después que se reconoció como elemento por Henry Cavendish (1731-1810),
otro físico y químico inglés, que lo aisló, recogiéndolo sobre mercurio, por primera vez
en 1766. Lo describió como un “inflammable air from metals”. Cavendish pensaba,
erróneamente, que su compuesto de origen era el metal y no el ácido. Una de las
primeras experiencias de generación de hidrógeno data del siglo XIX y corresponde a

2. INTRODUCCIÓN
13
uno de los padres de la química actual, Antoine Laurent de Lavoiser, que fue capaz de
su producción a partir de la disociación de la molécula del agua.
Las aplicaciones del hidrógeno a lo largo del tiempo han evolucionado desde su
uso en navegación, como gas de llenado de globos aerostáticos, a usos por la industria
farmacéutica, entre otros. El descubrimiento de la pila de combustible por William
Robert Grove en 1839, supuso un punto de inflexión en el uso del hidrógeno por su
capacidad de generar electricidad. Posteriormente, las exigencias técnicas de los viajes
espaciales de la NASA a mediados del siglo XX supusieron una mejora considerable en
los diseños de las pilas de combustible dada su capacidad de producir agua y
electricidad.
En la actualidad se produce a nivel mundial entre 40 y 45 millones de toneladas
de hidrógeno (más de 500.000 millones de metros cúbicos) mayoritariamente a partir de
gas natural. Aproximadamente dos terceras partes se usan en la industria química, para
la producción de amoniaco, y en las industrias de refino del petróleo. El resto del
hidrógeno producido se usa en otras aplicaciones industriales como: la fabricación de
vidrios, productos farmacéuticos, grasas, etc.
Los expertos coinciden en la importancia del hidrógeno como futura forma de
energía que paulatinamente sustituirá a los combustibles fósiles y cuya generación se
basará en las energías renovables logrando un sistema energético sostenible. Alcanzar
este horizonte no está exento de desafíos tecnológicos a los que se deberá enfrentar la
humanidad desarrollando la investigación en los métodos de producción y obtención del
hidrógeno así como en la aplicación y desarrollo de las pilas de combustible.
2.2.2 Importancia del hidrógeno
La reducción mundial de los problemas derivados del uso de combustibles
fósiles requiere un gran desarrollo tecnológico de alternativas y reestructuración de los
sistemas de energíaiix
. El Séptimo Programa Marco de la Comunidad Europea para
acciones de Investigación, Desarrollo Tecnológico y Demostración (2007-2013)
reconoce que no hay una única solución a los problemas energéticos, sino un amplio
abanico de tecnologías: energías renovables, captura y secuestro de dióxido de carbono,

2. INTRODUCCIÓN
14
biocarburantes económicamente viables…, y el uso de nuevos vectores de energía. En
este sentido, la Comunidad Europea apoya la intensificación de la investigación en este
ámbito, con vistas, en particular, a explorar nuevas soluciones asociadas a la utilización
del hidrógeno como vector energético alternativoiv,ix,
xiv,xv
.
El hidrógeno no se encuentra libre en la naturaleza, sino formando parte de
compuestos como los hidrocarburos y el agua. Por tanto, el hidrógeno puede ser
producido a partir de una de estas materias primas mediante aporte de energía.
Posteriormente, durante su consumo para producir energía, se libera en forma de agua,
sin producir ninguna otra emisión. De este modo, no puede ser destruido, al contrario
que los hidrocarburos, y simplemente cambia de estado iix
.
Por ello, se considera que el hidrógeno puede ser el vector energético del futuro
y su uso es una opción a largo plazo para reducir las emisiones ambientalesiix,xvi,xvii
. En
este contexto, nace la idea de “Economía del Hidrógeno”. Esto significa que el
hidrógeno sería utilizado para almacenar y transportar la energía producida por fuentes
renovables en lugares distantes, para abastecer energía en los puntos de consumo. Por
tanto, la combinación de las fuentes renovables para producir energía “limpia” y la
utilización de ésta (que no se puede almacenar) para generar hidrógeno, constituyen la
base de su desarrollo tecnológico.
Figura 2.1. Economía del Hidrógeno

2. INTRODUCCIÓN
15
2.2.3 Métodos para la producción de hidrógeno
El hidrógeno es un vector energético y puede producirse por una gran variedad
de métodos, cada uno de ellos se caracteriza por la fuente de energía primaria utilizada
para obtenerlo. Dentro de las fuentes primarias están incluidos el carbón, gas natural,
hidrocarburos, agua y alcoholes, y también se incluyen fuentes renovables como la
biomasa, la energía solar, la eólica, la hidráulica y la nuclearxviii
.
La elección del energético primario y la tecnología para producir hidrógeno
están sujetas a parámetros como el coste del ciclo de combustible y las influencias
externas, como los impactos ambientales y sociales. Las tecnologías de producción
presentan una gran cantidad de alternativas, existen procesos químicos, biológicos,
electrolíticos, fotolíticos y termoquímicos (reformado con vapor, oxidación parcial,
gasificación, electrólisis). Todas estas tecnologías se muestran en la Figura 2.2.
Figura 2.2. Producción de Hidrógeno a partir de diversas fuentes.
Todos estos métodos pasan por la obtención de gas de síntesis mediante alguno
de los siguientes procesos:

2. INTRODUCCIÓN
16
• Reformado con vapor de gas natural o naftas ligeras.
CH4 + H2O ↔ CO + 3H2CnHm + nH2O → nCO + (m/2+n) H2
• Oxidación de fracciones petrolíferas pesadas y (gasificación) de carbón y
biomasa.
CnHm + n/2 O2 → nCO + m/2 H2C + ½ O2
→ CO
Si el propósito es utilizar el gas de síntesis como materia prima para obtener
algún otro producto, sólo se necesita el reactor que produzca el reformado con vapor o
la oxidación parcial de la materia prima elegida. Si el objetivo es la producción de
amoníaco, se necesita un reformador secundario en el que se añade aire para completar
la conversión de metano, empleando el N2
aguas bajo para la síntesis de NH3. Si el
propósito es obtener H2
puro, se prescinde de este reactor. A continuación, se usan dos
reactores en serie para convertir el CO en CO2 e H2
a través de la reacción de
desplazamiento de gas de agua:
CO + H2O ↔ CO2 + H2
Posteriormente, transformar el CO remanente en metano (gas inerte para el
catalizador empleado en la síntesis de amoníaco) según la reacción de metanación:
CO + 3H2 ↔ CH4
+ H2O
Se están desarrollando alternativas a esta última etapa de purificación por
metanación, mediante PSA (adsorción por cambio de presión) o CO-PROX (oxidación
selectiva de CO). Por último, cabe mencionar que todos los procesos involucrados son
catalíticos. Actualmente, el 96% del hidrógeno se produce a partir de combustibles
fósiles, principalmente por reformado con vapor de metano (48%)xix,xx
. Sin embargo,
esta situación impediría alcanzar una economía del hidrógeno sostenibleixx
.
A continuación se expone en la Figura 2.3 las principales alternativas de
producción de hidrógeno, incluyendo tanto los procesos tradicionales, anteriormente
mencionados, como las nuevas tecnologías.

2. INTRODUCCIÓN
17
Figura 2.3. Producción de Hidrógeno puro.
2.2.3.1 Oxidación parcial de hidrocarburos
Se trata de un proceso exotérmico en el que un combustible reacciona con una
cantidad de oxígeno inferior a la necesaria para que se produzca la combustión completa
del mismo, obteniéndose hidrógeno. La reacción global se puede representar como:
CnHm + (n+a)/2O2
→ (n-a)CO + aCO2
+ (m/2)H2
Presenta la ventaja de que la tecnología utilizada para este proceso se encuentra
muy desarrollada, pero produce CO2, lo cual es un problema, puesto que las materias
primas de las que se parte suelen ser combustibles fósiles. Además, otro inconveniente
es su elevado coste de inversiónxx
.
Las conversiones que se alcanzan se encuentran entre el 70 y el 80 %.

2. INTRODUCCIÓN
18
2.2.3.2 Reformado con vapor de agua
El combustible reacciona con vapor de agua formando dióxido de carbono e
hidrógeno:
CnHm + n H2O → nCO + (m/2 + n) H2
CO + H2O ↔ CO2 + H2
CnHm + 2·n H2O → n CO2
+ (m/2 + 2·n) H2
Es una reacción endotérmica, por lo que es necesario un aporte suplementario de
calor para que tenga lugar la reacción, lo cual puede solventarse, sin afectar al buen
diseño ambiental, con el aporte de energía que provenga de fuentes renovables, o bien a
partir de otros combustibles, siempre que el procesamiento de los mismos incluya
mejoras medioambientalesxx
.
Al igual que en el caso de la oxidación parcial, este proceso cuenta con una
tecnología madura, pero la ventaja que presenta frente a la oxidación parcial es un
menor coste de inversión y una mayor conversión (superior al 90 %). Por otro lado,
también se presenta el inconveniente de la generación de dióxido de carbono, pero la
contribución al efecto invernadero y al calentamiento global dependerá del tipo de
materia prima que se utilice.
En este sentido, los alcoholes han demostrado buenas características para la
producción de hidrógeno al descomponerse fácilmente en presencia de agua por
reformado con vapor.
CH3OH + H2O → CO2 + 3H2
C2H5OH + 3H2O → 2 CO2 + 6 H2
El reformado con vapor de metanol ha sido ampliamente estudiado, pero el
principal inconveniente, junto con su relativamente alta toxicidad, es su origen a partir
de combustibles no renovablesxxi
. Sin embargo, el etanol puede ser muy utilizado por su
relativamente alto contenido en hidrógeno, buena disponibilidad y bajos costes de
producción, manipulación, transporte y almacenamiento fáciles y seguros, no ser tóxico

2. INTRODUCCIÓN
19
y la posibilidad de distribución en una red logística similar a las gasolineras
convencionales. Pero una de las razones más importantes para elegir etanol es que se
puede producir de manera renovable en grandes cantidades a partir de diversas fuentes
de biomasa (plantas de energía, residuos agrícolas de agro-industrias o materiales de
residuo forestal)xxii,xxiii
.
2.2.3.3 Reformado oxidativo con vapor de agua
El reformado catalítico de etanol con vapor es un proceso endotérmico donde el
calor de reacción debe ser suministrado por una fuente externa. Se trata de un sistema de
reacción muy complejo en el que muchas reacciones elementales son posibles. La
oxidación parcial de etanol tiene naturaleza exotérmica y, además, es muy rápida; sin
embargo, su rendimiento en hidrógeno es más bajo que en el reformado con vapor. La
energía que desprende la oxidación parcial sería la que necesita el reformado con vapor.
Los principales mecanismos de reacción implican reacciones de deshidratación o
deshidrogenación. Las reacciones de deshidratación producen productos intermedios
como el etileno, el cual se transforma fácilmente en carbono que se deposita sobre la
fase activa produciendo el envenenamiento del catalizador, disminuyendo la eficacia
hacia la producción de hidrógeno y reduciendo el tiempo de operación del catalizador.
Algunas rutas de reacción se pueden ver favorecidas según el catalizador
utilizadoxvi,xxii,xxiii,xxiv
.
El proceso de reformado oxidativo de etanol se rige por la siguiente ecuación
global:
C2H5OH + 1,8H2O + 0,6O2→ 2CO2 + 4,8H2
Se pueden diferenciar dos conjuntos de reacciones:
- Reacciones de descomposición de etanol:
C2H5OH → CO + CH4 + H2
C2H5OH → CH3CHO + H2

2. INTRODUCCIÓN
20
C2H5OH → C2H4 + H2O
C2H5OH + H2O → CO2 + CH4 + 2H2
- Reacciones de oxidación:
C2H5OH + O2 → CO2 + CO + 3H2
2C2H5OH + 5O2 4CO2 + 4H2O
2CH4 + 1,5O2 CO2 + CO + 4H2
CO + O,5O2 CO2
H2 + 0,5O2 H2O
2.2.3.4 Gasificación de biomasa con posterior conversión
En la misma línea de independizar la obtención del hidrógeno de las materias
primas fósiles, la biomasa, al ser renovable, es una de las fuentes más prometedoras.
Los estudios más avanzados se basan en su gasificación combinada con conversión
basada en la reacción de desplazamiento de monóxido de carbono (CO + H2O ↔ CO2 +
H2). Este planteamiento está en consonancia con las líneas de investigación potenciadas
por los gobiernos de las principales potencias mundiales. Así, en el Sexto Programa
Marco de la Unión Europea se fija como objetivo “el desarrollo de tecnologías eficaces
energética y económicas para la producción de gases ricos en hidrógeno a partir de
distintas biomasas, incluyendo los residuos procedentes de biomasas” (Comisión
Europea, 2003). Por su parte, el Departamento de Energía de Estados Unidos (DOE)
dentro del programa “Visión 21” está financiando proyectos dirigidos a la separación de
hidrógeno de mezclas de gases obtenidas en diferentes procesos industriales, siendo un
ejemplo de los mismos la gasificación.
Como la biomasa tiene un mayor contenido en volátiles (70-86% en base seca)
que el carbón (hasta un 30%), la primera etapa de pirólisis de la gasificación juega un
papel más importante con la biomasa. Se produce el craqueo térmico de la fase gaseosa
formada, reduciendo los niveles de alquitrán, que se gasifica en la segunda etapa del
proceso mediante reacciones con oxígeno, vapor e hidrógeno. Parte del alquitrán sin

2. INTRODUCCIÓN
21
convertir puede quemarse para liberar el calor necesario para las reacciones de pirólisis
endotérmicasxxv
.
2.2.3.5 Electrólisis del agua.
Un proceso alternativo que actualmente supone el 4% de la producción mundial
de hidrógeno es mediante electrolisis del agua. No obstante, se prevé un aumento
importante en la producción de hidrógeno por esta vía. Por tanto, es lógico intentar
desligar completamente el hidrógeno de dicho tipo de materias primas. Un factor a
considerar son los recientes desarrollos de aplicar energías baratas con la electrólisis del
agua, como la solarxxvi,xxvii,xxviii,xxix
.
Los equipos más habituales son los electrolizadores alcalinos, que emplean
como electrolito una disolución alcalina, típicamente disoluciones de hidróxido
potásico. Las reacciones que tienen lugar en estos sistemas son las siguientes:
Cátodo 2H2O → H2 + 2OH- - 2e
-
Ánodo 2OH- → 0,5O2 + H2O + 2 e
-
Célula H2O → H2 + 0,5O2
Las investigaciones sobre la electrolisis clásica se dirigen al desarrollo de
electrolizadores de halogenados y de membrana de intercambio protónicoxiix
. También
existen líneas de investigación sobre métodos electrolíticos no convencionales como la
electrolisis de vapor a alta temperatura (900-1.000 ºC). Este método tiene la ventaja de
que proporciona la energía de reacción necesaria en forma de calor y electricidad. Otras
investigaciones se dirigen a la electrolisis reversible del ácido bromhídrico. La energía
eléctrica necesaria para disociar esta molécula es la mitad que en el caso de la molécula
de agua. Una línea que está despertando especial interés en EE.UU. es la producción de
hidrógeno vía fotoelectroquímica. Este sistema es capaz de dividir la molécula de agua
en hidrógeno y oxígeno, usando sólo la luz solar. A diferencia de los sistemas
fotovoltaicos, éstos no necesitan cableado o convertidores externos. El sistema de

2. INTRODUCCIÓN
22
recolección de radiación solar es capaz de generar suficiente voltaje para descomponer
el agua.
• Electrólisis del agua utilizando energía solar.
El uso de energía solar para nuestras necesidades eléctricas cotidianas tiene
distintas ventajas: evitamos el consumo de recursos naturales y la degradación del
medio ambiente que resulta de las emisiones contaminantes, derrames de petróleo y los
productos tóxicos secundarios. Sin embargo, hay una desventaja en la energía solar, el
sol no brilla constantemente. Necesitamos, pues, un método para almacenar la energía
solar para utilizarla cuando no haya sol. El hidrógeno provee un método seguro,
eficiente y sano para hacerlo (Figura 2.4).
El ciclo del hidrógeno solar es un proceso en el que la electricidad producida por
los módulos solares opera un equipo de electrólisis que divide el agua (H2O) en sus
componentes elementales, hidrógeno (H2) y oxígeno (O2). El oxígeno se libera al aire y
el hidrógeno se bombea a los tanques, donde es almacenado en el lugar de producción o
se envía a las regiones donde el sol escasea. En la noche, cuando no se dispone de
energía solar, el hidrógeno se combina nuevamente con el oxígeno del aire en una celda
de combustible, y una planta de energía electroquímica convierte en electricidad la
energía química contenida en el hidrógeno. El único subproducto que resulta de este
proceso es agua pura.
Figura 2.4. Funcionamiento de una planta solarxxx
.

2. INTRODUCCIÓN
23
• Producción de hidrógeno mediante energía eólica.
Uno de los avances técnicos clave que hace esta perspectiva factible es el
almacenamiento en forma de hidrógeno de la energía producida por los
aerogeneradores. Hasta el momento, las grandes limitaciones de la energía eólica son su
variabilidad de acuerdo a las condiciones climáticas y su incapacidad de
almacenamiento. Sin embargo, esta tecnología permitirá gestionar y almacenar en forma
de energía química la energía eléctrica producida por los aerogeneradores.
El procedimiento es el siguiente, la energía eléctrica que se desea almacenar se
deriva hacia un electrolizador, que es un dispositivo en el que el paso de la corriente
disocia agua en sus dos componentes: oxígeno (O2) e hidrógeno (H2). Mientras que el
O2, que no tiene contenido energético, se libera a la atmósfera, el H2 obtenido se
comprime para hacer más fácil su almacenamiento en un volumen más pequeño, y se
mantiene almacenado en recipientes a presión hasta el momento en el que debe
emplearse para generar energía eléctrica en situaciones de demanda o necesidad de
gestión. En este caso, el hidrógeno se utiliza como carburante en un grupo de
generación eléctrica cuyo motor es similar a los de gas natural adaptado para hidrógeno.
Este motor aspira aire atmosférico cuyo oxígeno, en proporción del 20%, es el que,
provocado por la chispa de las bujías, reacciona con el hidrógeno en los cilindros. La
combustión del hidrógeno y del oxígeno libera sólo agua en un proceso inverso al que
se había producido en el electrolizador. Y el cigüeñal del motor arrastra un generador
que produce nuevamente energía eléctrica que se entrega a la red.
En general, este proceso es muy caro porque requiere de mucha energía y
presenta ciertas complicaciones técnicas. Sin embargo, la producción de hidrógeno con
los excedentes de la energía eólica es una solución sostenible. Básicamente, por la
variabilidad de la demanda energética: los aerogeneradores producen energía las 24
horas, pero la demanda de consumo es alta de día y baja por la noche, por lo que se
puede aprovechar esa producción para la elaboración de hidrógeno.
En este sentido, gracias al hidrógeno la energía eólica sí sería sostenible. Con el
desarrollo de esta nueva tecnología, se aprovecharía plenamente el potencial y la
inversión en los parques eólicos y se fortalecería su aporte en términos de la demanda
total de energía. De esta forma, no sólo se estaría disminuyendo la dependencia de los

2. INTRODUCCIÓN
24
combustibles fósiles y los niveles de contaminación por las emisiones de dióxido de
carbono, sino que haría sostenible el desarrollo sustentable. En la Figura 2.5 se
representa el esquema del funcionamiento de una estación eólica con todos sus
elementos hasta que llega a la red de suministro eléctrico.
Figura 2.5. Funcionamiento del almacenamiento de energía eólicaxxxi
.
2.2.3.6 Energía nuclear
En la actualidad se ha desarrollado un reactor VHTR (Very-High-Temperature
Reactor) que hace más viable la producción de hidrógeno mediante energía nuclear. Es
un reactor moderado por grafito y enfriado por helio con neutrones térmicos. Está
diseñado para máxima eficiencia y opera a 1.000 ºC. Puede proveer electricidad y
también calor para procesos de alta temperatura en la industria petroquímica y
producción de hidrógeno. El reactor de referencia es de 600 MW. Aún deben
desarrollarse soluciones adecuadas para el manejo de residuos nucleares. La tecnología
básica del VHTR proviene de los reactores a gas de alta temperatura desarrollados en
EEUU y Alemania, y además hay más prototipos en desarrollo en Sudáfrica, Japón,
Francia, Corea, EEUU y China. Sin embargo, la meta de operar sobre los 1.000 ºC,
implica desafíos significativos en términos de desarrollo de combustible y materiales,
así como en términos de seguridad. Se busca definir sus conceptos básicos para el 2010
y optimizarlos para el 2015. Este reactor proporciona calor a altas temperaturas por la
base del reactor que permite usos tales como producción del hidrógeno o calor del
proceso para la industria petroquímica u otras.

2. INTRODUCCIÓN
25
El sistema de VHTR se diseña para ser un sistema de alta eficiencia que puede
proveer calor de proceso a un amplio espectro de procesos de alta temperatura. El
sistema puede incorporar el equipo de generación de la electricidad para resolver
necesidades de la cogeneración. El sistema también tiene la flexibilidad de adoptar
ciclos del combustible del uranio/del plutonio y de ofrecer la minimización en gastos de
combustible. Así, el VHTR tiene unas características de seguridad deseables ofrecidas
por los reactores enfriados por gas de alta temperatura modulares.
Los objetivos del NGNP (Next Generation Nuclear Plant) son:
1. Demostrar la producción nuclear-asistida, segura y económica del hidrógeno y
de la electricidad.
2. Demostrar la base para la comercialización del sistema nuclear, de la facilidad
de producción del hidrógeno, y del concepto de la conversión de la energía.
3. Establecer la base para la normativa de la comisión reguladora nuclear de la
versión comercial de NGNP.
En la Figura 2.6 se puede observar un esquema de un reactor VHTR donde
aparece el reactor con el refrigerante de helio, el intercambiador de calor y la unidad de
producción de hidrógeno.
Figura 2.6. Reactor de alta temperatura y unidad de producción de hidrógenoxxxii
.

2. INTRODUCCIÓN
26
La Tabla 2.1 muestra una comparativa de los procesos de producción de
hidrógeno.
Tabla 2.1. Comparativa de los procesos de producción de hidrógeno.
PROCESO VENTAJAS INCONVENIENTES
Oxidación parcial Muy avanzada
Emisiones de CO2
Gran infraestructura
Elevados costes de
inversión
Reformado con
vapor
Alta eficiencia
Desarrollado a gran escala
H2 a bajo coste
Emisiones de CO2
Gran infraestructura
No comercial a pequeña
escala
Reformado
oxidativo con vapor
Muy avanzada
Alta eficiencia
H2 a bajo coste
Emisiones de CO2
Gran infraestructura
No comercial a pequeña
escala
Gasidicación
Desarrollo de la
tecnología
Materias primas
abundantes y a bajo coste
Emisiones de CO2 altas
Gran infraestructura
Eficiencia baja
Electrólisis
Emisiones de CO2 bajas
Tecnología probada
H2 de gran pureza
Elevados costes
energéticos
Energía nuclear Alta eficiencia
Gran infraestructura
No comercial a pequeña
escala
Residuos nucleares
2.2.4 Almacenamiento del hidrógeno
Para conseguir que se generalice el uso del hidrógeno como vector energético, se
debe lograr su transporte y almacenamiento de forma económica. Esto supone un
considerable cambio con respecto al transporte y almacenamiento de los combustibles
fósiles convencionales, debido a la baja densidad energética de este gas. En la
actualidad existen distintas formas de almacenar hidrógeno, tanto para aplicaciones
estacionarias como para el sector del transporte (en forma gaseosa, líquida, combinado
químicamente o adsorbido en sólidos porosos), dependiendo su elección de diferentes
factores como el proceso final en el que se vaya a emplear, la densidad energética
requerida, la cantidad a almacenar y la duración del almacenamiento, la existencia de

2. INTRODUCCIÓN
27
otras posibles formas de energía disponibles, los costes y necesidades de mantenimiento
de la instalación, y los costes de operaciónxxxiii
.
En el caso del empleo de hidrógeno como combustible para el transporte, uno de
los principales problemas a resolver es la falta de los medios adecuados para su
almacenamiento en el propio vehículo, cumpliendo los requisitos de seguridad, costes, y
las características de suministro requeridas. El Departamento de Energía de Estados
Unidos establece como objetivos a conseguir en el almacenamiento de hidrógeno al
menos una eficiencia en peso (relación entre el hidrógeno almacenado y el peso del
sistema de retención) del 6%, o expresado en densidad, 60 kg·m-3
ya que un vehículo
con una pila de combustible de hidrógeno necesitaría más de 3 kg de hidrógeno para
una autonomía de unos 500 kmxxxiv,xxxv
.
• Almacenamiento en forma gaseosa
Dado que el hidrógeno es producido en forma gaseosa y sus aplicaciones suelen
requerir que se encuentre en este estado, la vía más simple podría ser su
almacenamiento a alta presiónxxxvi
. Este tipo de almacenamiento (presiones superiores a
20 MPa) requiere que los depósitos sean pesados y voluminosos, además de plantear
cuestiones de seguridad tanto en los vehículos como en los depósitos de
almacenamiento, distribución y carga de hidrógeno.
Cuando se compara esta alternativa frente al empleo de otros combustibles, el
almacenamiento de hidrógeno gaseoso en recipientes a presión no resulta competitivo
debido a su baja densidad y al elevado coste de los recipientes a presión y del propio
proceso de compresión del hidrógenoxxxvii
.
• Almacenamiento en forma líquida
La opción del almacenamiento de hidrógeno en estado líquido en recipientes
criogénicos requiere alcanzar temperaturas de almacenamiento muy bajas (21,2 K),
haciendo inevitable su pérdida por volatilización incluso empleando las mejores
técnicas de aislamiento. Además, el alto consumo energético asociado al enfriamiento,
aproximadamente el 30% de la energía almacenada, hace que esta opción resulte
inviable en la práctica, desde el punto de vista económico, salvo en aquellas

2. INTRODUCCIÓN
28
aplicaciones donde el coste de hidrógeno no sea un factor crítico y éste sea consumido
en cortos periodos de tiempo (por ejemplo, en aplicaciones aeroespaciales)xxvi,xxxviii
.
• Combinación química (hidruros metálicos)
Numerosos metales de transición, y sus aleaciones, pueden ser utilizados para
almacenar hidrógeno en forma de hidruros metálicos. Estos hidruros se forman por
reacción con hidrógeno, siendo éste absorbido en la estructura metálica, y pudiendo ser
desorbido gracias a pequeñas variaciones de presiónxxv
.
Además de la dificultad que supone el intentar reducir la temperatura y presión
de desorción de los hidruros con mayor capacidad de almacenamiento de hidrógeno,
esta alternativa presenta un serio problema relacionado con el elevado peso del sistema
de almacenamiento como consecuencia de los bajos niveles de retención de hidrógeno
que se consiguen (<2% a temperaturas inferiores a 423 K)xxxix
.
• Adsorción en sólidos porosos (nanoestructuras de carbono)
Recientemente, se ha planteado la posibilidad de llevar a cabo el
almacenamiento de hidrógeno mediante adsorción en un sólido poroso, lo que
presentaría la ventaja de ser una forma más segura y sencilla de manejar el hidrógeno,
reduciéndose drásticamente la presión necesaria para su almacenamiento.
En este sentido, los primeros trabajos publicados, basados en nanoestructuras de
carbono cuyos resultados no fueron corroborados por ningún investigador, mostraban
almacenamientos excepcionales de hasta el 60% en peso. Desde entonces y hasta el
momento, se está dedicando un gran esfuerzo al estudio de nanoestructuras de carbono
con elevada superficie específica (fibras, nanotubos y carbones activos) concluyendo
que la cantidad de hidrógeno adsorbida a baja temperatura (77 K) es proporcional a la
superficie específica BET de la nanoestructura de carbono, independientemente de la
estructura geométrica del carbón, con valores máximos muy inferiores a los
anteriormente indicados. También se concluye que la cantidad de hidrógeno fisisorbido
a temperatura ambiente y presiones de hasta 35 MPa es inferior al 0,1% en peso para
cualquiera de las nanoestructuras estudiadas, lo que cuestiona su potencial utilidad para
esta aplicaciónxxix,xl
.

2. INTRODUCCIÓN
29
• Otros métodos en desarrollo
Actualmente, se están desarrollando diferentes actividades relacionadas con el
almacenamiento de hidrógeno en dos familias de nuevos materiales con potenciales
aplicaciones en procesos de separación y almacenamiento de hidrógeno; estructuras
organometálicas porosas isoreticulares (IRMOFs) y materiales organosilíceos
periódicos mesoestructurados (PMOs). También, se trabaja en la predicción de las
propiedades relacionadas con el almacenamiento de hidrógeno de estos materiales
mediante estudios de simulación molecular y su verificación mediante la obtención de
los datos experimentales correspondientes a la cinética y el equilibrio de adsorción de
hidrógeno.
2.2.5 Aplicaciones del hidrógeno
Los principales sectores de consumo actual de hidrógeno se muestran en la
Figura 2.7. Hoy en día, el hidrógeno se utiliza principalmente en la producción de
amoníaco, refino del petróleo y síntesis de metanol. También se utiliza en el programa
espacial de la NASA como combustible de las lanzaderas aeroespacialesvii
. Sin
embargo, aparte de las aplicaciones convencionales, es necesario destacar el papel que
las pilas de combustible jugarán en el consumo de hidrógeno en un futuro próximo
como elementos intermediarios para la generación de energía eléctrica “limpia”.
Figura 2.7.Distribución del consumo actual de Hidrógeno por aplicación.

2. INTRODUCCIÓN
30
Principales usos del hidrógeno:
• Industriales
Si bien en los últimos años el hidrógeno ha cobrado notoria relevancia como
combustible del futuro, sus aplicaciones en diversos sectores industriales son bien
conocidasxli,xlii
:
1. Industria Química: El hidrógeno es un compuesto de gran interés para la
industria química, participando en procesos de hidrogenación o como agente reductor en
procesos redox. A continuación se citan algunos de los más importantes:
Industria del refino: Los procesos de hidrogenación en refinería tienen
como objetivo principal la obtención de fracciones ligeras de crudo a partir de
fracciones pesadas, aumentando su contenido en hidrógeno y disminuyendo su peso
molecular. De forma simultánea pueden eliminarse elementos indeseables como azufre,
nitrógeno y metales, con objeto de cumplir la normativa en la formulación de los
distintos productos de la refinería.
Síntesis inorgánica. El hidrógeno es imprescindible en procesos de
importancia comercial como por ejemplo la producción de ácido clorhídrico, peróxido
de hidrógeno, hidroxilaminas, etc. Sin embargo, destaca de forma sobresaliente la
síntesis de amoníaco, para la cual es imprescindible el uso de H2, junto con el N2, en la
formación de dicha molécula, que posteriormente se empleará en la obtención de sales
de amonio para fertilizantes, ácido nítrico, nitratos, etc.
Síntesis orgánica. En química orgánica el hidrógeno participa en un gran
número de procesos de hidrogenación o reducción para la obtención de productos
químicos e intermedios. Destaca la utilización del denominado gas de síntesis, una
mezcla formada principalmente por CO y H2, para la obtención de otros muchos
productos químicos, principalmente metanol, pero también oxoalcoholes, isocianatos,
ácido acético, acetatos, combustibles sintéticos, metano, etileno, etc.
Otros: El hidrógeno también es materia prima o interviene en los
procesos de producción de productos químicos de uso cotidiano como pueden ser
detergentes, materiales poliméricos, productos intermedios del sector textil, etc.

2. INTRODUCCIÓN
31
2. Industria electrónica: El hidrógeno se usa para la fabricación de ciertos
componentes electrónicos. Por ejemplo, para producir semiconductores dopados, se
depositan en una matriz de silicio cantidades traza de elementos (Si, As, Ge, etc.) en
forma de hidruros, mezclados con una corriente de hidrógeno de elevada pureza.
3. Industria metalúrgica: El hidrógeno se utiliza en este sector industrial para
conseguir atmósferas anti-oxidantes, necesarias en ciertos procesos, o para tratamientos
térmicos. Se emplea como agente reductor para la producción de hierro (reducción
directa del mineral) y en procesos de producción de otros metales no-férricos (como
cobre, níquel, cobalto, molibdeno, uranio, etc.). También es habitual añadir diferentes
proporciones de hidrógeno a las corrientes gaseosas empleadas en diferentes procesos
de corte y soldadura, tratamientos superficiales (atomización) y tratamientos en
atmósferas especiales (templado, sinterización, fusión, flotación de vidrio, etc.).
4. Otras: En la industria del vidrio se utiliza para el pulido térmico del vidrio,
dando lugar a un acabado superficial excepcional. En la industria agroalimentaria el
hidrógeno es utilizado para la modificación de propiedades físico-químicas, tales como,
punto de fusión, estabilidad química y disminución del color y olor, en las grasas,
aceites y ácidos grasos.
• Energéticos
Como se mencionó con anterioridad, la relevancia que el hidrógeno ha
adquirido durante los últimos años viene dada por su utilización como combustiblexli,xlii
.
El hidrógeno puede quemarse directamente para la generación de electricidad mediante
turbinas de gas y ciclos combinados o utilizarse como combustible, tanto de motores de
combustión interna como en pilas de combustible. Las principales ventajas de este
compuesto se centran en las elevadas eficacias que pueden alcanzarse y en que el único
producto de su combustión es vapor de agua, sin generar NOX, si se controla la
temperatura para inhibir la reacción entre el nitrógeno y el oxígeno atmosféricos, y de
CO2, evitando la contribución al calentamiento global.
En relación con la utilización del hidrógeno como combustible, hay tres
aplicaciones posibles:

2. INTRODUCCIÓN
32
1. Combustión directa: La combustión del hidrógeno con oxígeno puro conduce
a la formación de vapor de agua puro: 2H2 + O2
→ 2H2O, obteniéndose una temperatura
de los gases superior a 3000 ºC en la zona de la llama. Sin embargo, esto conlleva
problemas con los materiales de los equipos empleados y la generación de NOx. Para
solventar estos inconvenientes, puede recurrirse a la inyección de agua en la corriente de
hidrógeno, lo que permite ajustar la temperatura del vapor al valor deseado, o bien al
empleo de catalizadores basados en platino, consiguiendo que la reacción tenga lugar a
temperaturas desde ambiente hasta 500 ºC. Los gases de combustión producidos pueden
llevarse directamente a una turbina de gas o a un ciclo combinado de turbina de
vapor/turbina de gas para la generación de electricidad.
2. Combustible en motores: Esta aplicación se encuentra tradicionalmente en su
empleo en la industria aeroespacial, como combustible de vehículos espaciales (además
de servir como suministro de energía para los ordenadores y sistemas de soporte en el
espacio), obteniendo agua como “subproducto”. Así, los programas espaciales son los
mayores consumidores de hidrógeno líquido, habiendo adquirido gran experiencia en su
manejo que puede ser la base de futuros desarrollos en otros campos. Sin embargo, esta
opción es también aplicable a vehículos de transporte por carretera. De hecho, las
investigaciones actuales se están centrando tanto en motores de combustión externa
(motores Stirling), como interna, para vehículos de transporte terrestre, aéreo y
marítimo. El uso de hidrógeno en motores de combustión interna es un campo que está
recibiendo cada vez más interés, siendo un 20% más eficaz que los que emplean
gasolina, debido a las características del hidrógeno (elevada difusividad, amplio
intervalo de inflamabilidad y alta temperatura de auto-ignición).
3. Pilas de combustible: La revolución energética que supone la economía del
hidrógeno se basa en el uso de este gas por medio de las llamadas pilas de combustible,
en las que se combina, por vía electroquímica, con el oxígeno para la producción de una
corriente eléctrica. Junto con la utilización de motores eléctricos y las baterías de nueva
generación serán los sustitutos de los actuales motores de combustión interna y las
instalaciones de combustión locales para el abastecimiento de necesidades tanto
estacionarias (domésticas e industriales) como móviles. Una pila de combustible
consiste en un ánodo en el que se inyecta el combustible (comúnmente hidrógeno) y un
cátodo en el que se introduce un oxidante (normalmente aire u oxígeno). Los dos

2. INTRODUCCIÓN
33
electrodos de una pila de combustible están separados por un electrolito conductor de
iones. Los reactivos se transforman electroquímicamente, de acuerdo con las
semirreacciones:
Ánodo: H2 → 2H
+ + 2e
-
Cátodo: 1/2 O2 + 2H
+ + 2e
- → H2O
Global: H2 + 1/2 O2
→ H2O + electricidad (E
o
= 1,23 V)
Se genera de esta forma una corriente eléctrica entre ambos electrodos que, a
diferencia de lo que ocurre en una pila o batería convencional, no se agota con el tiempo
de funcionamiento ni necesita ser recargada, sino que se prolonga mientras continúe el
suministro de los reactivos. Así pues, se transforma la energía química, almacenada en
el enlace H-H de la molécula H2, en energía eléctrica y vapor de agua (Figura 2.8). Esta
transformación utiliza directamente la energía libre disponible en el combustible a su
temperatura de operación y no está limitada por el ciclo de Carnot, alcanzando
rendimientos superiores a los procesos convencionales.
Figura 2.8. Esquema de funcionamiento de una pila de combustible de Hidrógeno-
Oxígeno.
Las pilas de combustible pueden ofrecer la respuesta a diversos requerimientos
energéticos. La eficacia de estos dispositivos no depende del tamaño como sucede en
otros sistemas energéticos. Este hecho permite su aplicación en sistemas de energía

2. INTRODUCCIÓN
34
miniaturizados y portátiles. Su eficacia es potencialmente superior a cualquier otro
sistema, haciéndolas particularmente atractivas para aplicaciones estáticas de alta o baja
energía. Además, las pilas de combustible suponen actualmente una esperanza real
dentro del mercado del transporte.
2.3 Bioetanol
2.3.1 Importancia actual y perspectiva
El bioetanol es el producto de fermentación alcohólica de diversos materiales
orgánicos a través de la acción de microorganismos. La producción de bioetanol perdió
importancia a finales de la primera mitad del siglo XX, al ser sustituida por la
producción de etanol por vía sintética, a partir de derivados del petróleo, que resulta más
barata, pero no puede ser utilizado en la preparación de alimentos, bebidas alcohólicas,
ni medicamentos.
La elevación de los precios del petróleo hicieron volver los ojos hacia la vía
fermentativa de producción de etanol, y hoy se trabaja fundamentalmente en la
búsqueda de materias primas baratas, que sustituyan a las tradicionales materias
azucaradas, como melazas, productos intermedios de la producción de azúcar y, jugos
de frutas, entre otros, a la vez que se busca una mayor eficiencia en los procesos de
fermentación y recuperación y purificación al alcohol producidoxliii
.
El coste de producción del etanol está íntimamente relacionado y es dependiente
del coste de la materia prima puesta en fábrica, del volumen y de la composición de la
misma. El éxito de cualquier plan de desarrollo de cultivos para la producción de etanol
es dependiente de la selección de los cultivos apropiados, los métodos de producción y
su ubicación. Un sistema que sea establecido alrededor de los costos más bajos de la
materia prima y esté completamente integrado, de forma tal que aproveche todas las
posibilidades que le dan los derivados, presenta las mejores oportunidades para ser
exitoso.
En la actualidad, la ruta preferida comercialmente para la producción de etanol
es la hidratación de etileno, el cual es producido mediante craqueo térmico de naftas.
Sin embargo, ante la necesidad de evitar el uso de derivados fósiles y la creciente

2. INTRODUCCIÓN
35
demanda de etanol como combustible, se busca la posibilidad de producir bioetanol en
elevadas cantidades a partir de la fermentación de carbohidratos de biomasa, tales como
azúcares, almidón o celulosa, procedentes de caña de azúcar, cereales, residuos
agrícolas o forestales e incluso basura orgánica urbana. Este medio de obtención es
interesante pues, tras la reacción para obtener hidrógeno, todo el CO2 generado cierra el
ciclo de carbono, pues es el mismo que los microorganismos necesitaron para fermentar
los azúcares a alcoholes, de forma que, con cada tonelada de etanol utilizada en
reemplazo de combustibles tradicionales la emisión de CO2
disminuye 2,3
toneladasxx,xli
.
En los países de la Organización para la Cooperación y el Desarrollo Económico
(OCDE), la mayoría del bioetanol es producido a partir del almidón de cosechas de
cereal. El maíz se usa principalmente en Estados Unidos y el trigo, la remolacha
azucarera y la cebada en Europa, donde cabe destacar que España lidera desde 2002 la
producción de bioetanol. En los procesos convencionales, sólo se utiliza la parte de
almidón de la planta y existen considerables residuos fibrosos en forma de cáscaras de
semilla y tallos, cuyo componente principal es celulosa. Sin embargo, aunque la
producción comercial actual de etanol a partir de biomasa de celulosa es baja, se está
realizando considerable investigación en este área en los países de la OCDE,
especialmente en Estados Unidos y Canadáii. El enfoque actual es optimizar el valor
total de las cosechas produciendo productos de mayor valor como alimento y fibra, y
usando “desechos” (residuos de biomasa) para obtener etanol y electricidad. De este
modo, se crea una sinergia en lugar de una competición entre las industrias alimentaria
y del combustible por la misma fuente de biomasa. Para comparar el residuo de la
biomasa y el crudo de petróleo en valores de energía, la producción de residuo anual de
biomasa mundial (desecho de madera, paja, cáscaras, vainas, tallos) iguala la mitad de
la producción anual de petróleoxx
.
2.4 Reacción Water Gas Shift
En la década de los 80 aumentó la preocupación por el calentamiento global y el
cambio climático, causados principalmente por las emisiones de CO2 a la atmósfera,
resultado del consumo de combustibles fósiles. Esta preocupación dio lugar al protocolo

2. INTRODUCCIÓN
36
de Kyoto cuya finalidad fue establecer unos objetivos de reducción de las emisiones de
CO2 antropogénico. Pero es bien sabido, que la presencia de estos combustibles sigue
siendo la primera fuente energética mundial. Para alcanzar los objetivos marcados por
este protocolo se debe:
Conseguir soluciones efectivas, a un coste asumible, que reduzcan de
forma considerable las emisiones de CO2 a la atmósfera.
Aumentar la proporción de energía renovable en el consumo de energía
primaria, convirtiéndose la biomasa en la principal protagonistaxliv
.
Una de las alternativas más estudiadas, consiste en la transformación de la
energía presente en los combustibles fósiles a hidrógeno. Esta transformación llevaría
consigo la producción de gas de síntesis, para su posterior transformación a hidrógeno
y CO2, y finalmente a la necesaria separación de los productos de reacción por medio de
sistemas de membranas. El desarrollo de esta tecnología puede jugar un importante
papel en las técnicas de producción de hidrógeno, ya que consigue la captura de CO2 de
forma económicamente viable.
La tecnología más extendida para la producción de CO2 e H2 a partir de gas de
síntesis es el reformado con vapor de agua. Este reformado con vapor puede llevase a
cabo mediante multitud de alternativas. Estas abarcan desde los reactores de lecho fijo,
o de membrana, a incluso, la utilización de bacterias en los procesos denominados
biológicos.
La reacción WGS es la que consigue la transformación de CO a CO2
produciendo hidrógeno, mediante reformado con vapor de agua. Esta tecnología viene
siendo utilizada desde hace tiempo, la primera referencia bibliografía de esta reacción es
de Mond L. y Langer C. en 1888, por lo que es una tecnología madura, tanto a escala de
laboratorio como a escala planta piloto.
La reacción es un equilibrio catalítico, exotérmico, y limitado
termodinámicamente. La estequiometria del equilibrio es la siguiente:
CO + H2O CO2 +H2 ΔH= - 41,1 KJ/mol

2. INTRODUCCIÓN
37
Al aumentar la cantidad de agua, el equilibrio se desplazará hacia la derecha,
aumentando la conversión de CO. Mientras que un aumento de la temperatura tiene el
efecto contrario disminuyendo la conversión al afectar a la velocidad de reacción.
Debido al hecho de que la reacción es exotérmica, la velocidad de reacción disminuye al
aumentar la temperatura, por lo que será preciso un enfriamiento óptimo en el momento
en el que la reacción llega al equilibrio.
2.4.1 Aplicaciones de la reacción Water Gas Shift
Las aplicaciones de esta reacción WGS se pueden estudiar desde el punto de
vista energético, debido la generación de H2 y desde el punto de vista del medio
ambiente, debido a la conversión de grandes cantidades de CO hacia CO2.
Con esta tecnología se puede conseguir que las emisiones de CO2 de las
centrales térmicas dejases de ser un problema, consiguiendo el secuestro de éste
posteriormente a la conversión de CO por WGS.
La aplicación energética está relacionada con las pilas de combustible y el
hidrógeno generado como producto de la reacción de WGS. La producción de
hidrógeno se consigue a partir de gas de síntesis de un GICC (ciclo combinado
integrado de gasificación). Separando el CO2 y el H2 obtenido en la reacción, pueden
utilizarse para la obtención de energía eléctrica en pilas de combustible con
rendimientos teóricos superiores al 80%.
En resumen, la tecnología de Water Gas Shitf, asegura mejoras directas en la
producción de energía eléctrica y mejoras inducidas al permitir potenciar el uso del
hidrógeno como vector energético limpio en sustitución de otros agentes, contribuyendo
por ello a un balance ambiental global muy positivo. Por otra parte, se podría reducir la
cantidad de CO2 emitido a la atmósfera, debido a las posibilidades a medio plazo de
secuestro de CO2 capturándose de forma selectiva al H2.
De esta forma se desarrolla desde el proyecto SINGULAR el estudio preliminar
para la construcción de una planta piloto de captura, generación y almacenamiento de
CO2 utilizando un 2% en volumen del gas de síntesis generado en Elcogas.

3 OBJETIVOS Y ALCANCE DEL PROYECTO

3. OBJETIVOS Y ALCANCE DEL PROYECTO
39
El principal objetivo de la presente investigación es evaluar cuales son las
mejores condiciones y el catalizador de mayores garantías en las reacciones de
reformado oxidativo de etanol con vapor y Water Gas Shift. Su finalidad es maximizar
la producción de H2 obteniendo una selectividad y una conversión de etanol elevados.
Cabe destacar el papel vital que tiene la elección del catalizador para ambos procesos.
El programa de investigación planteado incluye las siguientes etapas:
1. Síntesis de 4 catalizadores con fase activa de Ni soportados sobre YSZ
(10% Ni, 15% Ni y 23%Ni).
2. Caracterización de los catalizadores de Ni-YSZ utilizados para el
reformado, y del catalizador comercial de CrO3 y Fe2O3 utilizado para la
reacción WGS.
Análisis por difracción de rayos X (DRX).
Análisis de espectroscopía de emisión atómica de plasma acoplado por
inducción (ICP-AES).
Análisis de temperatura programada de reducción (TPR).

3. OBJETIVOS Y ALCANCE DEL PROYECTO
40
Análisis de adsorción-desorción de N2 a 77 K para obtener la superficie
específica, tamaño y distribución de poro.
3. Ensayos catalíticos con un lecho (reformado) y dos lechos (reformado +
WGS). La finalidad del doble lecho es aprovechar el CO producido en el
primer lecho, para convertirlo en H2 y aumentar el rendimiento del proceso
global.
4. Análisis de resultados y conclusiones.

4 METODOLOGÍA EXPERIMENTAL

4. METODOLOGÍA EXPERIMENTAL
42
4.1 Preparación de los catalizadores
4.1.1 Síntesis del soporte
Para la obtención de los catalizadores se utilizó como YSZ. La primera etapa era
de desgasificación. Se añadía el soporte a un matraz de fondo redondo y se mantenía a
vacío con ayuda de un rotavapor (Figura 4.1), a una temperatura de 40ºC y una
velocidad de giro media. La duración de esta etapa era de dos horas.
Figura 4.1. Imagen del rotavapor.

4. METODOLOGÍA EXPERIMENTAL
43
4.1.2 Impregnación
Se preparaba previamente la disolución de catalizador con nitrato de níquel
disuelto en agua [(NO3)2·6H2O]. Con ayuda de una pipeta Pasteur se añadía gota a gota
dicha disolución al soporte desgasificado, procurando la máxima distribución.
Posteriormente se volvía a utilizar el rotavapor, en esta ocasión a una temperatura de
90ºC y velocidad de giro media. Esta etapa de impregnación duraba dos horas, aunque
este tiempo era variable dependiendo de la cantidad de fase activa añadida al soporte, ya
que cuanto mayor era la fase activa a incorporar, mayor era el volumen de líquido a
utilizar y, por tanto, mayor su volumen a retirar.
4.1.3 Calcinación
Una vez acabada la última etapa en el rotavapor, se extraía el catalizador del
matraz de fondo redondo con ayuda de una espátula y se molía para asegurar un tamaño
de partícula uniforme. Se traspasaba a un vaso cerámico y se metía en la estufa durante
8 horas para eliminar los grupos nitrato del precursor. Después se introducía en la mufla
(Figura 4.2) aplicando una rampa de temperatura, desde la temperatura ambiente hasta
550ºC a una velocidad de 2ºC por minuto, manteniéndose esta temperatura máxima
durante 3 horas.
Figura 4.2. Imagen de la mufla programable.

4. METODOLOGÍA EXPERIMENTAL
44
4.2 Técnicas de caracterización
Los catalizadores sintetizados en la presente investigación han sido
caracterizados mediante diversas técnicas de análisis para la determinación de sus
principales propiedades físico-químicas. Las técnicas empleadas se describen
brevemente a continuación, detallándose los equipos empleados y las condiciones de
análisis.
4.2.1 Espectroscopía de emisión atómica de plasma acoplado por inducción (ICP-
AES)
Mediante esta técnica espectroscópica se determinó la composición química del
material sintetizado. En concreto, se utilizó para calcular el porcentaje en peso de los
distintos elementos incorporados en los catalizadores (níquel). El equipo utilizado para
realizar los análisis es un espectrofotómetro VARIAN Liberty RL Sequential ICP-AES
de análisis multielemental (Figura 4.3).
La espectroscopía de emisión atómica se fundamenta en la excitación de los
átomos metálicos mediante un plasma de argón, capaz de alcanzar 10000 K, asegurando
la completa atomización de la muestra en estado líquido. Al cesar la excitación, tiene
lugar la emisión de radiación por parte del metal para volver al estado energético
fundamental. La intensidad de dicha emisión permite cuantificar la concentración del
elemento ya que depende de la cantidad de átomos del mismo. La ventaja principal del
plasma es la alta temperatura alcanzable, que asegura la completa atomización de la
muestra cuando se sitúa en el plasma.
Previamente al análisis, las muestras sólidas fueron sometidas a un tratamiento
de digestión ácida con ácido clorhídrico, peróxido de hidrógeno y fluorhídrico para
conseguir la disolución de los metales. Para determinar la concentración de los metales,
es necesario obtener previamente las curvas de calibración correspondientes a cada
metal en el intervalo de concentración adecuado. Para cada metal se realizan cinco
puntos de calibración. Las disoluciones se preparan a partir de disoluciones patrón
certificadas para análisis de emisión atómica de 1000 mg/L en medio ácido nítrico.

4. METODOLOGÍA EXPERIMENTAL
45
Figura 4.3. Imagen del espectrofotómetro.
4.2.2 Difracción de rayos X (DRX)
Esta técnica se aplicó con el objetivo de la identificación de las fases metálicas
cristalinas soportadas. Los difractogramas de rayos X de los catalizadores se realizaron
en un difractómetro de polvo PHILIPS modelo X´PERT MPD equipado con
monocromador secundario de grafito (Figura 4.4), empleando radiación Kα del Cu
(1,54056 Å) con una intensidad de 40 mA y un potencial de 40 kV.
Figura 4.4. Imagen del difractómetro.

4. METODOLOGÍA EXPERIMENTAL
46
La difracción de rayos X aporta información directa de la estructura ordenada de
los materiales. Los rayos X son una radiación de longitud de onda comprendida entre
10-3
y 100 Å producida por el frenado de electrones de elevada energía o por
transiciones electrónicas entre niveles atómicos internos. Cuando los rayos X son
dispersados por un entorno ordenado, tiene lugar la difracción, debido a que las
distancias entre los centros de dispersión son del mismo orden que la longitud de onda
de la radiación incidente. La difracción se produce por efecto acumulativo de la
dispersión generada cuando el haz de RX interacciona con las diferentes capas
ordenadas que se encuentran a la misma distancia. Para observar la difracción, se
requiere que los centros de dispersión estén distribuidos en el espacio de manera
regular, formando planos con orientaciones específicas. Además, es necesaria una
distancia similar entre los planos responsables de la difracción que constituyen una
familia de planos. Así, cada familia de planos con la misma orientación espacial da
lugar a una señal de difracción si se cumple la Ley de Bragg:
n·λ = 2·dhkl·sen θ
donde n es el orden de difracción, λ es la longitud de onda de la radiación incidente,
dhkl es la distancia interplanar correspondiente a cada familia de planos denotadas por
los índices de Miller correspondientes (h, k, l) y θ es el ángulo de difracción. La
intensidad de los haces reflejados a cada ángulo se denomina difractograma y, a partir
de él, pueden determinarse las distancias atómicas interplanares características de el
sólido analizado.
Por otro lado, la desviación con respecto a las condiciones ideales de la ley de
Bragg (haz de RX paralelo y estrictamente monocromático), hace que el efecto de
difracción se produzca en un intervalo angular más o menos amplio, originando un
ensanchamiento de las líneas del difractograma alrededor del valor teórico de θ. Esta
anchura adicional (b) es debida tanto a factores instrumentales como al grado de
perfección cristalina de la muestra. Además, el tamaño excesivamente pequeño de los
cristalitos también provoca un ensanchamiento de los picos (β), de manera que cuanto
menor es el tamaño de los cristales, mayor es la anchura del pico de difracción. De esta
forma, la anchura total del pico obtenido en el difractograma estará integrada por ambas
contribuciones: anchura instrumental y ensanchamiento por tamaño pequeño de cristal.

4. METODOLOGÍA EXPERIMENTAL
47
La anchura instrumental (b) se determina midiendo la anchura del máximo de
difracción de una sustancia patrón bien cristalizado y cuyos cristales tengan un tamaño
medio lo suficientemente grande, para evitar el ensanchamiento debido a este efecto. En
esta investigación, se utilizó un patrón de calcita con tamaño de cristal superior a 1 μm.
La relación entre el ensanchamiento de los picos (β) y el tamaño de los cristales
de la muestra a analizar viene dada por la ecuación de Scherrer:
β=0,9*λ
𝐷ℎ𝑘𝑙 ∗ cos 𝜃
Donde Dhkl es la dimensión media de los cristales en la dirección normal a los
planos (hkl) que producen la difracción.
Finalmente, se requiere fijar la relación entre la contribución instrumental y la
debida al tamaño de cristal. Scherrer propuso una relación lineal:
W1/2 = b + β,
Siendo W1/2 la anchura del pico medida en el punto medio de la vertical trazada
en el máximo de intensidad.
4.2.3 Reducción térmica programada con hidrógeno (TPR)
Esta técnica se utilizó para conocer la reducibilidad de los catalizadores y, de
forma indirecta, obtener información sobre la fortaleza de las interacciones metal-
soporte e incluso sobre el grado de dispersión de la fase metálica. Los análisis de TPR
se llevaron a cabo en un equipo MICROMERITICS modelo AUTOCHEM HP con
detector de conductividad térmica (TCD) (Figura 4.5).

4. METODOLOGÍA EXPERIMENTAL
48
Figura 4.5. Imagen del modelo AUTOCHEM HP.
La reducción a temperatura programada es una técnica extremadamente sensible,
que permite estudiar el proceso de reducción de un catalizador o precursor reducible al
exponerlo a un flujo de una mezcla gaseosa reductora (típicamente un pequeño %vol de
H2 en un gas inerte), mientras se aumenta la temperatura linealmente. El principio de
operación de la técnica de TPR se basa en el cambio químico que experimenta un
sistema redox cuando se expone a un ambiente reductor. Dado que los materiales a
caracterizar son óxidos, en presencia de H2 se reducen para obtener el correspondiente
metal según la reacción:
MOx + xH2
→ M
0
+ xH2O
El proceso de reducción se sigue continuamente por medida de la composición
(contenido en H2) de la mezcla gaseosa reductora a la salida del reactor.
Para la realización de los análisis, en primer lugar, se coloca el sólido en un
reactor tubular de lecho fijo fabricado en cuarzo y, previamente al análisis, se
desgasifica para eliminar las sustancias adsorbidas físicamente. Posteriormente, se
enfria la muestra y se procede a la realización del análisis, haciendo pasar una corriente
de gas reductor (17 %vol de H2 en Ar, 100 mL/min) con un calentamiento hasta 900 ºC
a una velocidad linealmente programada de 5 ºC/min. Antes de alcanzar el detector,
para eliminar el agua formada y poder realizar el seguimiento de la cantidad de H2
consumido en cada momento, el gas efluente se hace pasar a través de una trampa fría,

4. METODOLOGÍA EXPERIMENTAL
49
consistente en una mezcla frigorífica de isopropanol y nitrógeno líquido a una
temperatura de -80 ºC.
4.2.4 Adsorción-desorción de nitrógeno a 77 K
La determinación de las propiedades texturales de todos los soportes y
catalizadores sintetizados se realizó a partir de las isotermas obtenidas en un equipo de
adsorción volumétrico MICROMERITICS ASAP 2010 a la temperatura del nitrógeno
líquido (Figura 4.6).
Figura 4.6. Imagen del equipo MICROMETRIC ASAP 2010.
Esta técnica se basa en la adsorción-desorción física de gases (adsorbatos) en
sólidos (adsorbentes). Cuando una cierta cantidad de adsorbente se pone en contacto
con un volumen dado de una mezcla gaseosa que contiene el soluto a adsorber, se
produce la retención de soluto en la superficie del sólido acompañada de una
disminución de la concentración del mismo en la mezcla, hasta alcanzar el equilibrio de
adsorción. El adsorbato retenido, puede ser posteriormente desorbido del adsorbente por
una corriente de gas caliente o por reducción de presión. Si se mantiene constante la
temperatura, la relación entre la cantidad de soluto adsorbida y la concentración en la
disolución se denomina isoterma de adsorción-desorción. Dicha isoterma puede
determinarse de manera volumétrica, calculando la cantidad adsorbida mediante la

4. METODOLOGÍA EXPERIMENTAL
50
aplicación de las leyes de los gases a la presión y volumen de adsorbato antes y después
de la adsorción-desorción.
Mediante las isotermas adsorción y desorción de un gas inerte en la superficie
del material y el tratamiento matemático con diferentes modelos de adsorción, se
pueden estimar parámetros texturales tales como la superficie específica, el volumen de
poros, el diámetro de poro medio, etc.
Previamente al análisis, las muestras se desgasificaron en flujo de nitrógeno
mediante diferentes programas de temperatura en función del tipo de material a analizar.
Una vez desgasificadas las muestras, la adsorción se realiza añadiendo cantidades
crecientes de nitrógeno, para abarcar todo el intervalo de presiones relativas hasta
aproximarse a la saturación (P/P0 = 0,995). Una vez alcanzada la saturación, la
desorción se lleva a cabo por vacío, reduciendo la presión relativa escalonadamente. El
tiempo de estabilización de cada medida se fijó en 5 s.
La estimación de la superficie específica se realiza utilizando el modelo
Brunauer Emmett-Teller (BET)xlv
aplicado a la rama de adsorción de nitrógeno en el
intervalo de presiones parciales seleccionado para cada catalizador, de forma que no se
produzca en ningún momento condensación capilar en mesoporos. Aunque la validez de
esta ecuación para materiales microporosos es cuestionablexlvi
estos valores pueden ser
utilizados razonablemente a efectos comparativos.
La estimación del tamaño de poro se ha realizado aplicando el modelo Barret-
Joyner-Halenda (BJH)xlvii
con la ecuación de Harkins - Jura para determinar el espesor
de la capa de nitrógeno adsorbida. Este modelo se aplica utilizando la rama de adsorción
y suponiendo una geometría cilíndrica de los poros.
4.3 Instalación experimental
La parte experimental se divide en tres secciones diferenciadas: alimentación al
sistema, reacción y análisis de productos. La Figura 4.7 representa el esquema de la
instalación en ensayos catalíticos con un lecho y la Figura 4.8 representa el esquema en
el caso de la adición del lecho WGS.
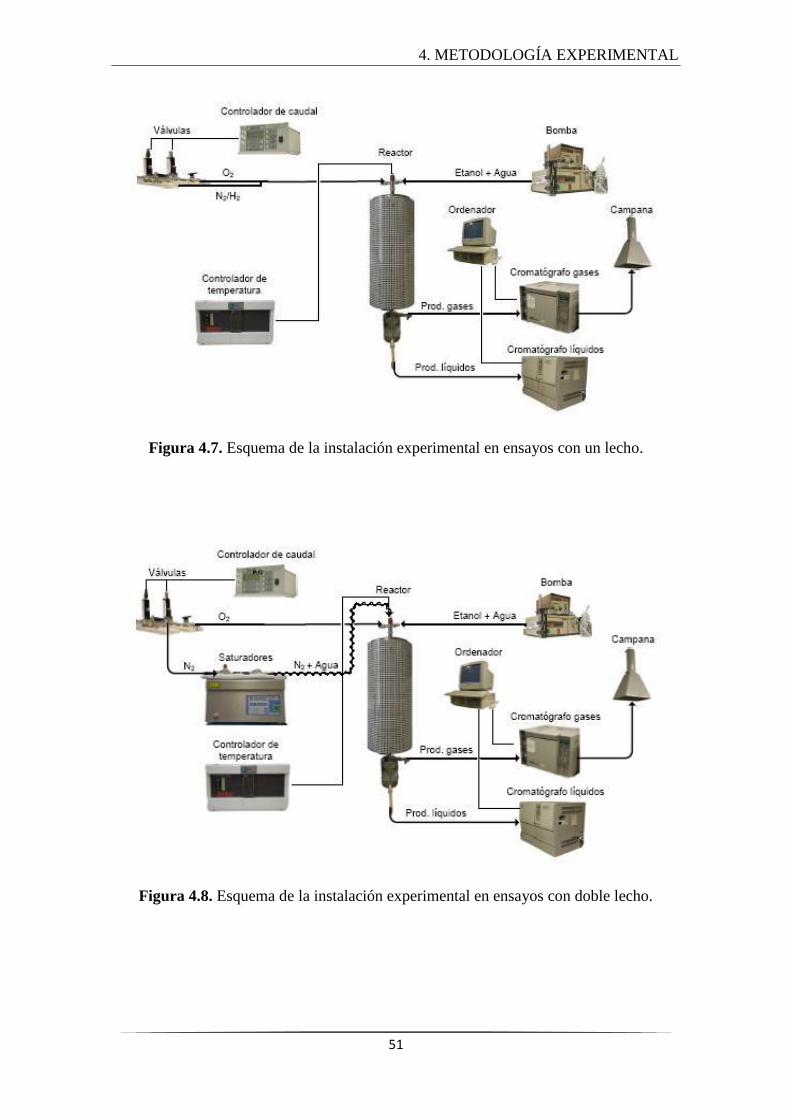
4. METODOLOGÍA EXPERIMENTAL
51
Figura 4.7. Esquema de la instalación experimental en ensayos con un lecho.
Figura 4.8. Esquema de la instalación experimental en ensayos con doble lecho.

4. METODOLOGÍA EXPERIMENTAL
52
4.3.1 Sección de alimentación
El sistema de alimentación está constituido por dos líneas de flujo continuo de
gases y una línea de flujo continuo de líquido. Por una de las líneas de gases circula el
H2 necesario para la reducción del catalizador utilizado en el reformado y, compartiendo
la misma línea, circula el N2 utilizado para el proceso de inertización del reactor y para
su posterior utilización durante la reacción, como compuesto estándar para el análisis de
productos y como gas portador del agua en las reacciones de doble lecho. Por la otra
línea circula el O2 necesario para la reacción. Por la línea de líquido circula una
disolución acuosa de etanol impulsada por una bomba GILSON 302. En el caso de los
ensayos catalíticos con doble lecho, el nitrógeno arrastra el agua contenida en dos
saturadotes dentro de un baño termoestático y es alimentada al reactor. Las
conducciones son de acero inoxidable de diámetro de 1/8 de pulgada. Las tres
conducciones poseen una válvula antirretorno (VR) para impedir la posible inversión
de flujo.
Los gases son controlados por dos controladores másicos, de la marca Brooks
Instruments (modelo 5850). Estos están constituidos por un sensor de conductividad
térmica, un indicador controlador y una electroválvula. Estos controladores fueron
calibrados con un caudalímetro de burbuja (HUMONICS, modelo Optiflow 520).
4.3.2 Sección de reacción
El sistema de síntesis estaba constituido por un reactor tubular de lecho fijo y
flujo descendente construido en cuarzo, de 25 mm de diámetro interno y 76 cm de
longitud (Figura 17). En la parte superior se insertaba lateralmente la conducción de
entrada de los gases reaccionantes y la conducción de entrada de la disolución de etanol
en agua. En el caso del doble lecho, el agua arrastrada por el nitrógeno es introducida a
la sección de bolas de cuarzo que separa los dos lechos, a través de una tubería de acero,
con el fin de alimentar sólo el lecho de WGS y desplazar el equilibrio a la producción
de hidrógeno.
Disponía de otro acceso vertical que permitía introducir un termopar (TI) para
medir la temperatura del lecho de reacción. Este termopar estaba conectado al

4. METODOLOGÍA EXPERIMENTAL
53
controlador de temperatura de un horno eléctrico LENTON THERMAL DESIGN, que
permitía monitorizar y controlar la temperatura en el reactor, así como realizar rampas
de calentamiento y enfriamiento. De este modo, se conseguía la eliminación de
gradientes radiales de temperatura y oscilaciones de temperatura en el lecho catalítico
inferiores a 2 ºC.
El catalizador, soportado sobre una pequeña capa de lana de cuarzo, se situaba
en la parte media del reactor. La parte superior del mismo, libre de catalizador, servía
para la recalefacción de los reactivos. En la parte inferior del reactor se encontraba la
salida de los productos que eran posteriormente analizados.
Figura 4.9. Imagen del sistema de reacción.
4.3.3 Sección de análisis de productos
A la salida del reactor condensaba el líquido en un primer condensador enfriado
por hielo. Los gases de salida eran conducidos a un segundo condensador enfriado por
etilenglicol. Después pasaban por un filtro de gases y, finalmente, llegaban al
cromatógrafo. Toda la conducción desde la salida del reactor estaba traceada con hilo
calefactor.

4. METODOLOGÍA EXPERIMENTAL
54
El cromatógrafo de gases utilizado es un HEWLETT PACKARD MODELO
5890 (Figura 4.10). Éstos eran inyectados mediante una jeringa de muestreo, a 145ºC
utilizando Argón como gas portador eran analizados mediante un detector de
conductividad térmica (TCD). El líquido de reacción condensado a la salida del reactor
era analizado por inyección mediante una microjeringa utilizando un Espectrómetro de
masas en un cromatógrafo SHIMADZU GC-17A (Figura 4.11).
Figura 4.10. Imagen del cromatógrafo HEWLETT PACKARD MODELO 5890.
Figura 4.11. Imagen del espectrómetro de masas en un cromatógrafo SHIMADZU GC-
17A.
4.3.4 Parámetros de reacción
Los resultados catalíticos se expresaron a través de una serie de parámetros de
reacción en los cuales se basará la discusión de estos ensayos. A continuación, se define
cada uno de los parámetros, indicándose las ecuaciones utilizadas para su cálculo:

4. METODOLOGÍA EXPERIMENTAL
55
- Los valores de conversión de reactivos, expresados en porcentaje molar, se calcularon
como:
100
ientrada
isalidaientrada
reactivoF
FFX
donde Fi representaba el caudal molar del componente i.
- Los valores de las selectividades hacia los diferentes productos expresados en
porcentaje molar, se calcularon con la siguiente expresión:
100ducidostotalespro
producto
productoF
FS
Otro parámetro interesante es la relación CO2/COx, calculada como:
COCO
CO
COCOSS
SR
x
2
2
2 /
4.4 Productos químicos utilizados
Los productos comerciales utilizados en la presente investigación se enumeran a
continuación según su aplicación, y se especifica la casa comercial suministradora.
Síntesis de catalizadores
- Nitrato de níquel (II) hexahidratado, Ni(NO3)2·6H2O 97%, (PANREAC).
- YSZ (IONOTEC).
- Catalizador comercial Fe,Cr (RESEARCH AND CONSULTING)
Reducción
- Hidrógeno, 99,998 %, (PRAXAIR).
Inertización y gas de arrastre

4. METODOLOGÍA EXPERIMENTAL
56
- Nitrógeno, 99,999 %, (AIR LIQUID).
Reacción
- Oxígeno, 99,999%, (PRAXAIR).
- Agua desionizada.
- Etanol (C2H5OH) grado HPLC 99,9%, (VWR).
Gas portador en el cromatógrafo
- Argón, pureza del 99,996 %, (PRAXAIR).
Caracterización
- Nitrógeno líquido (Air Liquide).
- 2-Propanol, pureza del 99,8%, (SIGMA ALDRICH).
4.5 Procedimiento experimental
Previamente a los ensayos catalíticos se procedía a la activación del catalizador
de reformado, por reducción del metal a la fase activa Ni0. Para ello, se situaba en el
lecho catalítico una cantidad determinada del mismo, y se alimentaba una corriente de
H2 (100 mL/min) a 400 ºC durante 2 h. Posteriormente se hacía pasar una corriente de
N2 para inertizar el medio de reacción y eliminar los restos de H2. En los ensayos
catalíticos con doble lecho, el catalizador de WGS no necesitaba ser reducido debido a
que su fase oxidada es la activa.
Los ensayos catalíticos se llevaron a cabo a una presión de 2 bares y a una
temperatura de 400ºC. Se introducía un caudal de 46 ml/min de N2, 26.5 ml/min de O2 y
16,09·10-2
ml/min de mezcla alimento de agua/etanol con relación molar 1,8. El baño
termostático se mantenía a 82ºC para asegurarse una saturación adecuada que
introdujese al medio de reacción el suficiente agua para lograr una relación H2O/CO de
4-5.

4. METODOLOGÍA EXPERIMENTAL
57
Durante los experimentos se realizaba el análisis de las corrientes de salida cada
20 minutos. Los efluentes gaseosos se introducían en el cromatógrafo mediante una
jeringa. Los efluentes condensados se recogían para el análisis de la cantidad de agua y
compuestos orgánicos. El tiempo de reacción era de tres horas.
Una vez acabado el ensayo se procedía a la limpieza del reactor. Se extraía el
catalizador, la lana de cuarzo y parte del coque formado. Después se colocaba de nuevo
el reactor y se introducía O2 a 800ºC para la combustión total del coque. Una vez limpio
se cargaba de nuevo para el siguiente ensayo catalítico.

5 RESULTADOS Y DISCUSIÓN

5. RESULTADOS Y DISCUSIÓN
59
5.1 Caracterización del soporte YSZ
5.1.1 Difracción de rayos X (DRX)
La difracción de rayos X se realizó como prueba cualitativa para verificar la
estructura del soporte. La Figura 5.1 muestra el difractograma de DRX de la YSZ.
Figura 5.1. Difractograma del soporte YSZ.
El análisis de YSZ muestra sus picos característicos a 30º, 35º, 50º, 60º y 74º.

5. RESULTADOS Y DISCUSIÓN
60
5.1.2 Adsorción-desorción de N2
En la Figura 5.2 se muestra la isoterma de adsorción-desorción de nitrógeno a 77
K del soporte YSZ, junto con su distribución de tamaño de poro correspondiente.
Figura 5.2. Isoterma de adsorción-desorción y distribución de tamaño de poro del
soporte YSZ.
La isoterma de la YSZ es de tipo III según la clasificación de la IUPAC
correspondiente a materiales macroporosos en los que el llenado de los macroporos
tiene lugar por condensación capilar. En las isotermas se distinguen tres regiones:
adsorción monocapa-multicapa, condensación capilar y adsorción en multicapas sobre
la superficie externa de las partículas.
El cambio de pendiente debido a la condensación capilar es indicativo de un
llenado de poros a partir del cual se puede estimar el diámetro de los mismosxlviii
. Una
distribución de tamaño de poro más estrecha indica un mayor ordenamiento de la
estructura porosa. La YSZ muestra un sistema de poros poco homogéneo.
Los valores de las propiedades texturales se resumen en la Tabla 5.1
Tabla 5.1. Propiedades texturales de los soportes.
Soporte Propiedades texturales
Area BET (m2/g) Dporo (Å) Vporo (cm
3/g)
YSZ 13,3 74,24 2,6 E-2

5. RESULTADOS Y DISCUSIÓN
61
5.2 Caracterización de catalizadores
5.2.1 Espectroscopía de emisión atómica de plasma acoplado por inducción (ICP-
AES)
En la Tabla 5.2 se muestra el contenido real de metal medido con esta técnica.
Tabla 5.2. Contenido metálico de los catalizadores.
Catalizador % Fase activa
YSZ 10% Ni 9,2
YSZ 15% Ni 15,5
YSZ 23% Ni 23
WGS comercial Fe 52
Cr 4
5.2.2 Difracción de rayos X (DRX)
A partir del análisis por rayos X de los catalizadores (Figura 5.3, Figura 5.4 y
Figura 5.5) se observó que todas las muestras presentaron los picos de difracción
correspondientes al NiO a valores de 2θ = 37.3, 43.3, 62.9 y 75,4, y después de
reducirlas presentaron sus picos de difracción característicos del Ni a valores de 2θ =
44.5, 51.8 y 76.1.

5. RESULTADOS Y DISCUSIÓN
62
0 20 40 60 80
0
200
400
600
800
1000
NiO
NiO NiO
Ni NiNi
Ni
NiOIn
tensid
ad (
u.a
.)
2 (º)
Figura 5.3. Difractograma del catalizador YSZ-10%Ni.
0 20 40 60 80
0
100
200
300
400
500
600
700
NiO
NiO
NiO
Ni
Ni
Ni
Inte
nsid
ad (
u.a
.)
2 (º)
Figura 5.4. Difractograma del catalizador YSZ-15%Ni.
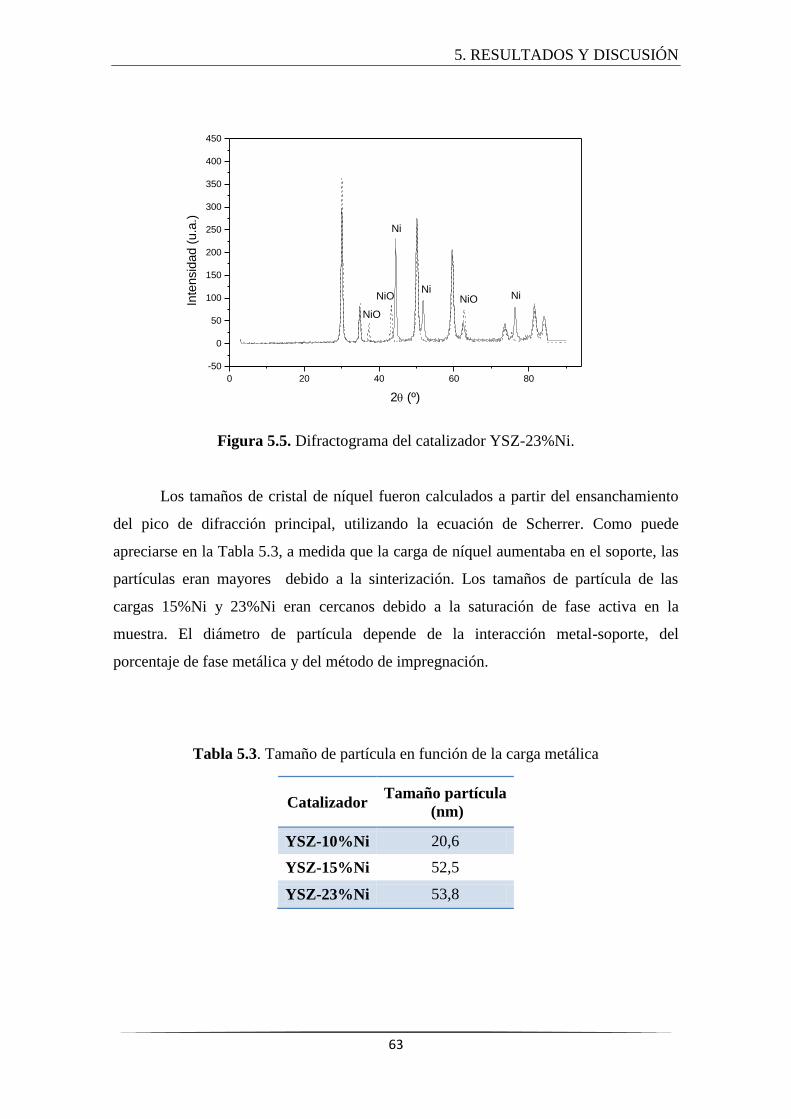
5. RESULTADOS Y DISCUSIÓN
63
0 20 40 60 80
-50
0
50
100
150
200
250
300
350
400
450
NiO
NiO NiONi
Ni
Ni
Inte
nsid
ad (
u.a
.)
2 (º)
Figura 5.5. Difractograma del catalizador YSZ-23%Ni.
Los tamaños de cristal de níquel fueron calculados a partir del ensanchamiento
del pico de difracción principal, utilizando la ecuación de Scherrer. Como puede
apreciarse en la Tabla 5.3, a medida que la carga de níquel aumentaba en el soporte, las
partículas eran mayores debido a la sinterización. Los tamaños de partícula de las
cargas 15%Ni y 23%Ni eran cercanos debido a la saturación de fase activa en la
muestra. El diámetro de partícula depende de la interacción metal-soporte, del
porcentaje de fase metálica y del método de impregnación.
Tabla 5.3. Tamaño de partícula en función de la carga metálica
Catalizador Tamaño partícula
(nm)
YSZ-10%Ni 20,6
YSZ-15%Ni 52,5
YSZ-23%Ni 53,8

5. RESULTADOS Y DISCUSIÓN
64
El difractograma del catalizador comercial WGS muestra los picos
característicos de las fases activas de Fe2O3 (2θ = 25, 54 y 62) y Cr2O3 (2θ = 28, 33 y
36). Al ser un catalizador comercial, el resto de picos se atribuyen principalmente al
soporte y a otras especies metálicas de menos relevancia (Figura 5.6).
0 20 40 60 80
0
20
40
60
80
100
120
140
Cr2O
3
Cr2O
3
Cr2O
3
Fe2O
3
Fe2O
3
Fe2O
3
Inte
nsid
ad
(u
.a.)
2 (º)
Figura 5.6. Digractograma del catalizador comercial WGS.
5.2.3 Adsorción-desorción de N2
En las Figura 5.7, Figura 5.8 y Figura 5.9 se muestran las isotermas obtenidas
para todos los catalizadores, junto con la distribución de tamaño de poros. Se puede
observar que todos los catalizadores de Ni-YSZ presentan una forma muy similar a la
del soporte YSZ, lo que implica que la estructura se ha mantenido como se había
detectado con el análisis por DRX.
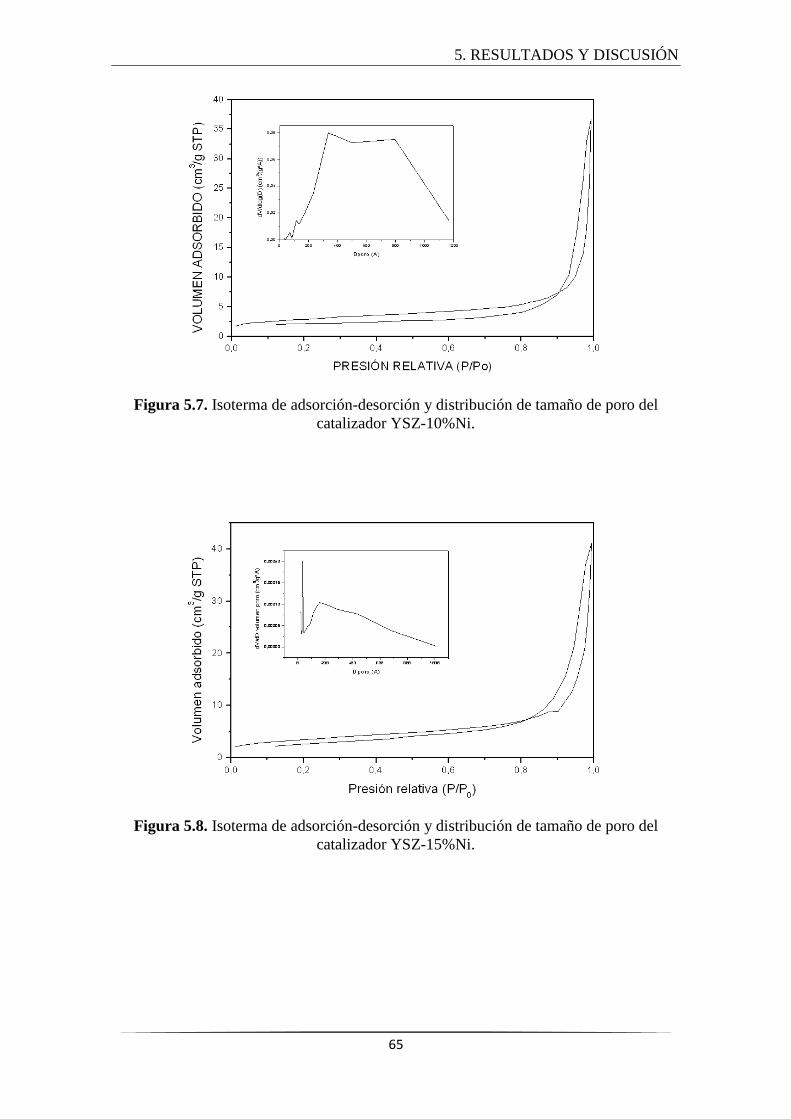
5. RESULTADOS Y DISCUSIÓN
65
Figura 5.7. Isoterma de adsorción-desorción y distribución de tamaño de poro del
catalizador YSZ-10%Ni.
Figura 5.8. Isoterma de adsorción-desorción y distribución de tamaño de poro del
catalizador YSZ-15%Ni.
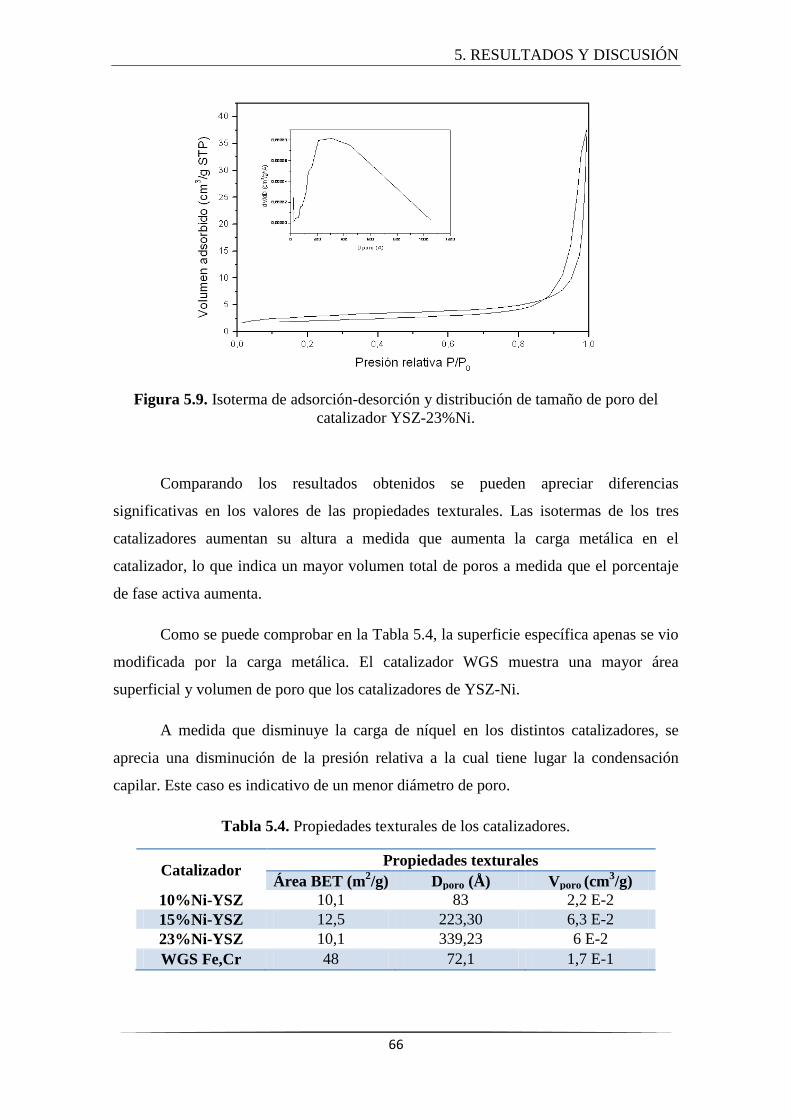
5. RESULTADOS Y DISCUSIÓN
66
Figura 5.9. Isoterma de adsorción-desorción y distribución de tamaño de poro del
catalizador YSZ-23%Ni.
Comparando los resultados obtenidos se pueden apreciar diferencias
significativas en los valores de las propiedades texturales. Las isotermas de los tres
catalizadores aumentan su altura a medida que aumenta la carga metálica en el
catalizador, lo que indica un mayor volumen total de poros a medida que el porcentaje
de fase activa aumenta.
Como se puede comprobar en la Tabla 5.4, la superficie específica apenas se vio
modificada por la carga metálica. El catalizador WGS muestra una mayor área
superficial y volumen de poro que los catalizadores de YSZ-Ni.
A medida que disminuye la carga de níquel en los distintos catalizadores, se
aprecia una disminución de la presión relativa a la cual tiene lugar la condensación
capilar. Este caso es indicativo de un menor diámetro de poro.
Tabla 5.4. Propiedades texturales de los catalizadores.
Catalizador Propiedades texturales
Área BET (m2/g) Dporo (Å) Vporo (cm
3/g)
10%Ni-YSZ 10,1 83 2,2 E-2
15%Ni-YSZ 12,5 223,30 6,3 E-2
23%Ni-YSZ 10,1 339,23 6 E-2
WGS Fe,Cr 48 72,1 1,7 E-1

5. RESULTADOS Y DISCUSIÓN
67
Para todos los catalizadores sintetizados, tras la incorporación de la carga
metálica, el volumen de poro y el diámetro de poro aumentaba, debido a la acidez de la
disolución de impregnación del metal, que atacaba la estructura del soporte haciendo
que los valores de las propiedades texturales aumentasenxlix
.
5.2.4 Reducción térmica programada con hidrógeno (TPR)
Las Figura 5.10 y Figura 5.11 muestran los perfiles TPR de los catalizadores. A
partir de estos análisis se pueden determinar diferencias en la reducibilidad de cada uno,
que dependerá de la localización y dispersión del metal y de la interacción metal-
soporte.
0 200 400 600 800
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
15%Ni-YSZ
23%Ni-YSZ
10%Ni-YSZ
Inte
nsid
ad s
eñal (u
.a.)
T (ºC)
Figura 5.10. Ensayo de reducción térmica programada de los catalizadores 10, 15 y
23% Ni-YSZ.

5. RESULTADOS Y DISCUSIÓN
68
0 200 400 600 800 1000
0,10
0,15
0,20
0,25
0,30
0,35
0,40
FeO a FeFe
3O
4 a FeO
Fe2O
3 a Fe
3O
4
CrO3 a Cr
2O
3
Se
ña
l re
lativa
(u
.a.)
Tª (ºC)
Figura 5.11. Ensayo de reducción térmica programada del catalizador comercial WGS.
Los ensayos realizados a los catalizadores de YSZ muestran el pico de reducción
de NiO a Ni metálico. El perfil TPR del catalizador de WGS muestra dos picos bien
diferenciados debidos a la reducción de CrO3 a Cr2O3 y de Fe2O3 a Fe3O4. A altas
temperaturas se obtuvo un solapamiento de los picos correspondientes a la reducción de
Fe3O4 a FeO y de FeO a Fel.
En los perfiles de reducción se puede observar a qué temperatura aparecen las
máximas de reducción. Los resultados obtenidos se muestran en la Tabla 5.5.
Tabla 5.5. Temperaturas máximas de reducción.
Catalizador Temperatura [ºC]
YSZ 10% Ni 339
YSZ 15% Ni 327
YSZ 23% Ni 314
WGS comercial
Fe3-Fe2 228
Fe2-Fe 320
CrO-Cr 703

5. RESULTADOS Y DISCUSIÓN
69
Para un mismo soporte, la temperatura de reducción de los catalizadores es
menor para aquel con mayor carga metálica. Al haber más cantidad de Ni, la dispersión
será menor y, por lo tanto, las especies metálicas serán más fáciles de reducir al
disminuir la interacción entre el soporte y éstasli, lii, liii
.
5.3 Ensayos catalíticos
De los ensayos catalíticos se obtuvieron una serie de datos, que fueron
ordenados para la realización de los cálculos pertinentes para la obtención de los
principales parámetros de reacción (conversión, selectividad, relación CO2/CO y
relación H2/CO) y su posterior interpretación.
5.3.1 Efecto de la temperatura
Con objeto de conocer la variación de la distribución de los productos con
respecto a la temperatura, se realizaron diferentes estudios partiendo de una temperatura
de 300ºC. Los valores calculados de selectividad de los productos se muestran en la
Tabla 5.6.
Tabla 5.6. Selectividad [%] de productos de reacción.
Temperatura
[ºC] Hidrógeno Metano
Dióxido de
carbono
Monóxido
de
carbono
Acetaldehído Acetona
300 44,04 7,79 19,92 23,09 0 0
350 49,80 8,94 15,75 24,48 0 0
400 50,01 8,98 14,87 24,22 0,16 0,05
Tabla 5.6 (Cont.). Selectividad [%] de productos de reacción.
Temperatura [ºC] Ciclopentano Dietoxietano Dietoximetano Isohexano Heptano
300 0,80 3,69 0,48 0 0
350 0,52 0 0 0,38 0,01
400 0,72 0,8 0 0 0

5. RESULTADOS Y DISCUSIÓN
70
De los ensayos catalíticos se apreció como al aumentar la temperatura se
incrementa la selectividad de hidrógeno. También aumenta la selectividad de monóxido
de carbono, mientras que las selectividades de metano y dióxido de carbono
disminuyen. Las selectividades de todos los gases fueron muy similares a las
temperaturas de 350ºC y 400ºC, razón por la que no se realizaron ensayos a
temperaturas mayores.
La relación CO2/CO es importante de cuantificar debido a que una de las
intenciones del reformado oxidativo es la conversión total de CO a CO2. Un valor
elevado de esta relación indica una mayor conversión. La Figura 5.12 muestra los
valores obtenidos.
Figura 5.12. Relación CO2/CO (efecto de la temperatura).
A medida que disminuye la temperatura de la reacción se observa que también
disminuye la relación CO2/CO, lo que indica que aumenta la cantidad de CO en función
de la del CO2.
La conversión indica el grado de transformación del reactivo clave en los
ensayos catalíticos. En todos los ensayos realizados los valores de conversión de etanol
obtenidos fueron prácticamente totales como se aprecia en la Tabla 5.7.
Tabla 5.7. Conversión del etanol.
Temperatura
[ºC] [%]
300 99,34
350 99,77
400 99,62
0,0
0,1
0,2
0,3
0,4
0,5
300ºC 350ºC 400ºC
Re
laci
ón
CO
2/C
O

5. RESULTADOS Y DISCUSIÓN
71
La razón de que distintas temperaturas den lugar a diferentes selectividades se
debe, a que al tratarse de un proceso complejo con múltiples reacciones de reformado y
oxidación, unas son favorecidas sobre otras. Al aumentar la temperatura de reacción
mejora la selectividad de hidrogeno y monóxido de carbono. Esto se debe a que a estas
condiciones favorecen las dos principales reacciones de descomposición de etanol y de
reformado de vapor de agua. A bajas temperaturas de reacción se favorece la
deshidratación de etanol a etileno, el cual da lugar a la formación de coque.
La disminución de dióxido de carbono y metano con el incremento de la
temperatura es debido a la reacción WGS (desplazado el equilibrio hacia la producción
de hidrógeno), el reformado de metano y el reformado seco.liv
5.3.2 Efecto del tamaño de partícula
Con objeto de conocer la variación de la distribución de los productos con
respecto al tamaño de partícula de la fase activa de níquel, se realizaron ensayos
catalíticos con distinta carga metálica. Los valores calculados de selectividad de los
productos se muestran en la Tabla 5.8.
Tabla 5.8. Selectividad [%] de los productos de reacción (efecto del tamaño de
partículas de níquel).
Tamaño
partícula
(nm)
Hidrógeno Metano Dióxido de
carbono
Monóxido de
carbono Acetaldehído Acetona
20,6 50,01 8,89 14,87 24,22 0,16 0,05
52,5 46,04 10,49 13,76 27,56 0,1 0
53,8 40,47 16,12 4,53 35,57 0 0
Tabla 5.8 (Cont.). Selectividad [%] de los productos de reacción (efecto del tamaño de
partículas de níquel).
Tamaño partícula
(nm) Ciclopentano Dietoxietano
20,6 0,72 0,8
52,5 0,95 0,85
53,8 1,29 1,74

5. RESULTADOS Y DISCUSIÓN
72
En la Figura 5.13 se muestra la relación CO2/CO en función del tamaño de
partículas de níquel.
Figura 5.13. Relación CO2/CO (efecto del tamaño de partículas de níquel).
La conversión de etanol fue prácticamente completa en los tres ensayos, como se
puede apreciar en la Tabla 5.9.
Tabla 5.9. Conversión de etanol (efecto del tamaño de partículas de níquel).
Tamaño partícula
(nm) [%]
20,6 99,62
52,5 99,04
53,8 98,6
A menores tamaños de partículas de níquel se favorece las reacciones de
reformado y oxidación que dan lugar a la producción de hidrógeno. Para tamaños de
partículas más pequeños se incrementa la producción de CO2 frente a la de CO. Los
motivos principales de la tendencia apreciada están actualmente en estudio y no se
encontraron referencias bibliográficas con qué compararlo.
5.3.3 Estudio de doble lecho
Una vez optimizada la temperatura de reacción y el tamaño de partícula de la
fase activa de níquel, se planteó mejorar el rendimiento del proceso por medio de la
0
0,1
0,2
0,3
0,4
20,6 nm 52,5 nm 53,8 nm
Re
laci
ón
CO
2/C
O
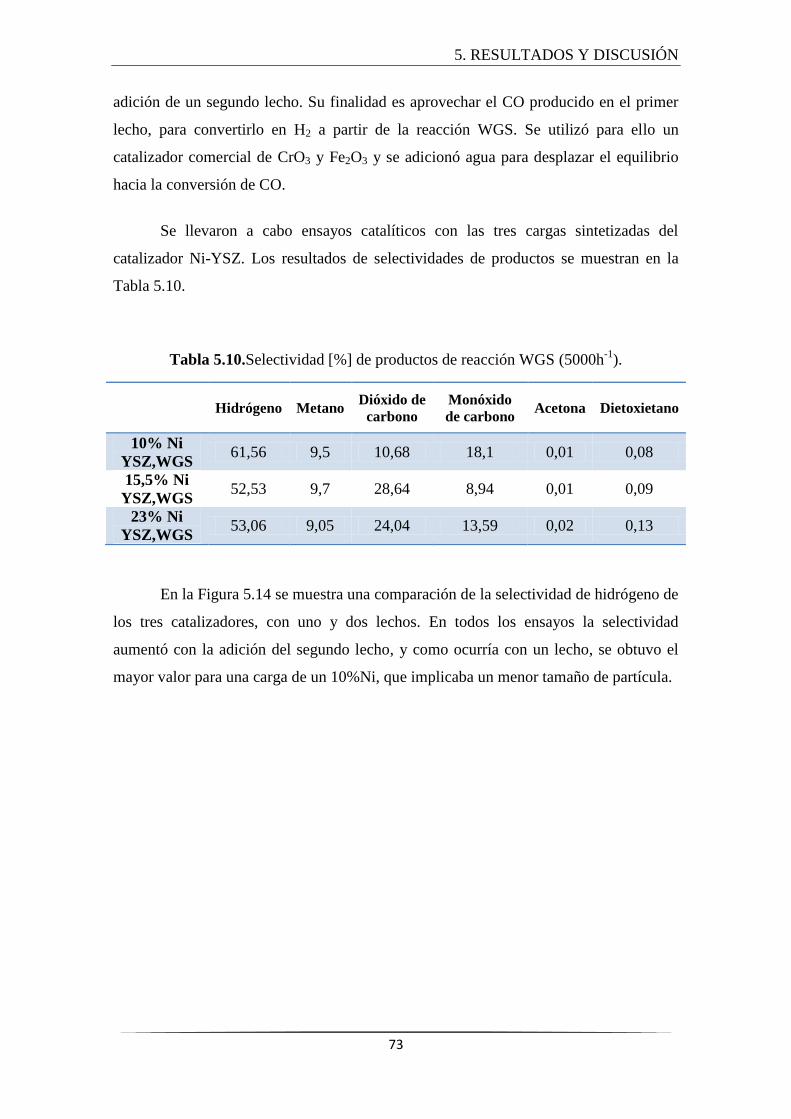
5. RESULTADOS Y DISCUSIÓN
73
adición de un segundo lecho. Su finalidad es aprovechar el CO producido en el primer
lecho, para convertirlo en H2 a partir de la reacción WGS. Se utilizó para ello un
catalizador comercial de CrO3 y Fe2O3 y se adicionó agua para desplazar el equilibrio
hacia la conversión de CO.
Se llevaron a cabo ensayos catalíticos con las tres cargas sintetizadas del
catalizador Ni-YSZ. Los resultados de selectividades de productos se muestran en la
Tabla 5.10.
Tabla 5.10.Selectividad [%] de productos de reacción WGS (5000h-1
).
Hidrógeno Metano Dióxido de
carbono
Monóxido
de carbono Acetona Dietoxietano
10% Ni
YSZ,WGS 61,56 9,5 10,68 18,1 0,01 0,08
15,5% Ni
YSZ,WGS 52,53 9,7 28,64 8,94 0,01 0,09
23% Ni
YSZ,WGS 53,06 9,05 24,04 13,59 0,02 0,13
En la Figura 5.14 se muestra una comparación de la selectividad de hidrógeno de
los tres catalizadores, con uno y dos lechos. En todos los ensayos la selectividad
aumentó con la adición del segundo lecho, y como ocurría con un lecho, se obtuvo el
mayor valor para una carga de un 10%Ni, que implicaba un menor tamaño de partícula.

5. RESULTADOS Y DISCUSIÓN
74
Figura 5.14. Selectividad de hidrógeno en uno y dos lechos (efecto del tamaño de
partículas de níquel).
En la Figura 5.15 se muestra una comparación de la relación H2/CO de los tres
catalizadores, con uno y dos lechos. En todos los ensayos la relación aumentó con la
adición del segundo lecho debido a la conversión del monóxido de carbono.
Figura 5.15. Relación H2/CO en uno y dos lechos (efecto del tamaño de partículas de
níquel).
0
10
20
30
40
50
60
70
10% Ni-YSZ, WGS 5000h
15% Ni-YSZ, WGS 5000h
23% Ni-YSZ, WGS 5000h
Sele
ctiv
idad
(%
)
lecho simple lecho doble
0
1
2
3
4
5
6
10% Ni-YSZ, WGS 5000h^-1
15% Ni-YSZ, WGS 5000h^-1
23% Ni-YSZ, WGS 5000h^-1
Re
laci
ón
H2
/CO
1 lecho 2 lechos

5. RESULTADOS Y DISCUSIÓN
75
Como puede apreciarse en la Figura 5.16, la relación CO2/CO aumenta en gran
medida con la adición de un segundo lecho con velocidad espacial de 5000h-1
. Esto se
debe que al haber más centro activos de CrO3 y Fe2O3, se favorece más el equilibrio
WGS, y por la adición de H2O se desplaza hacia la conversión de CO.
Figura 5.16. Relación CO2/CO en uno y dos lechos (efecto del tamaño de partículas de
níquel).
5.3.3.1 Efecto de la velocidad espacial
Se llevaron a cabo dos ensayos catalíticos con distinta velocidad espacial. Los
resultados de las selectividades se muestran en la Tabla 5.11.
Tabla 5.11. Selectividad [%] de productos de reacción (efecto de la velocidad espacial).
Hidrógeno Metano Dióxido de
carbono
Monóxido de
carbono Dietoxietano
WGS
10000h-1
55,5 13,87 13,36 15,86 0,35
WGS
5000h-1
63,09 8,92 12,61 15,39 0
0,00
0,20
0,40
0,60
0,80
10% Ni-YSZ, WGS 5000h
15% Ni-YSZ, WGS 5000h
23% Ni-YSZ, WGS 5000h
Re
laci
ón
CO
2/C
O
Lecho simple Lecho doble

5. RESULTADOS Y DISCUSIÓN
76
En la Figura 5.17 se muestra la comparación entre las selectividades de los
productos gaseosos en los ensayos catalíticos de YSZ-10%Ni con un lecho y doble
lecho a 5000 h-1
y 1000 h-1
.
Figura 5.17. Comparación de la selectividad de gases (efecto de la velocidad espacial).
El equilibrio WGS se favoreció hacia la conversión de CO para la producción de
H2 debido al agua alimentada al reactor. Se puede apreciar como a menor velocidad
espacial, la selectividad de hidrógeno es mayor.
En la Figura 5.18 se comparan los valores obtenidos de la relación CO2/CO,
entre los ensayos catalíticos con YSZ-10%Ni de un lecho y doble lecho (velocidad
espacial 10000h-1
y 5000h-1
del catalizador WGS).
0
10
20
30
40
50
60
70
Hidrógeno Metano Dióxido de Carbono
Monóxido de Carbono
Sele
ctiv
idad
(%
)
WGS 10000[1/h] WGS 5000[1/h] YSZ Ni 10%
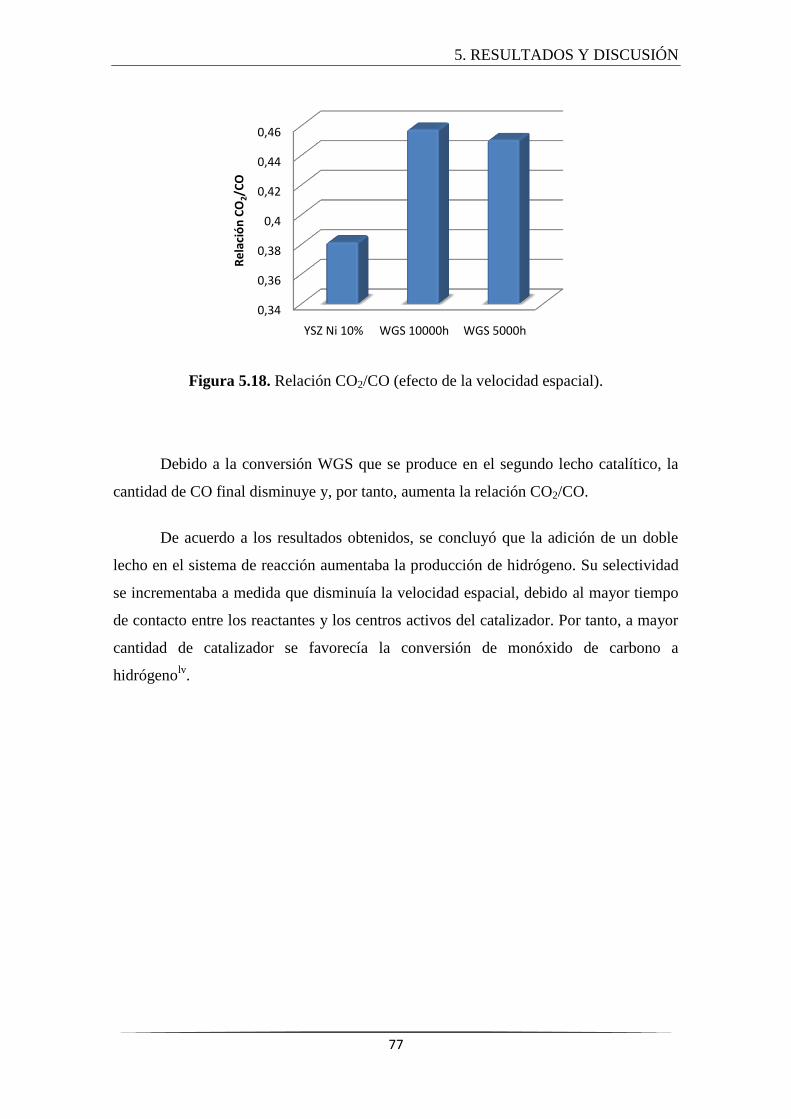
5. RESULTADOS Y DISCUSIÓN
77
Figura 5.18. Relación CO2/CO (efecto de la velocidad espacial).
Debido a la conversión WGS que se produce en el segundo lecho catalítico, la
cantidad de CO final disminuye y, por tanto, aumenta la relación CO2/CO.
De acuerdo a los resultados obtenidos, se concluyó que la adición de un doble
lecho en el sistema de reacción aumentaba la producción de hidrógeno. Su selectividad
se incrementaba a medida que disminuía la velocidad espacial, debido al mayor tiempo
de contacto entre los reactantes y los centros activos del catalizador. Por tanto, a mayor
cantidad de catalizador se favorecía la conversión de monóxido de carbono a
hidrógenolv
.
0,34
0,36
0,38
0,4
0,42
0,44
0,46
YSZ Ni 10% WGS 10000h WGS 5000h
Re
laci
ón
CO
2/C
O

6 CONCLUSIONES

6. CONCLUSIONES
79
A continuación, se resumen las principales conclusiones que se pueden extraer
de los resultados obtenidos en el transcurso de la presente investigación:
1. La adición de mayor carga metálica al catalizador Ni-YSZ suponía una
impregnación de partículas de níquel de mayor tamaño.
2. Para un mismo soporte, la temperatura de reducción de los catalizadores
era menor para aquel con mayor carga metálica. Al haber más cantidad
de Ni, la dispersión era menor y, por lo tanto, las especies metálicas eran
más fáciles de reducir al disminuir la interacción entre el soporte y éstas.
3. El proceso de reformado oxidativo de etanol ofreció conversiones muy
elevadas en todos los casos estudiados de temperaturas y tamaños de las
partículas de níquel.
4. A partir de 400ºC, los resultados obtenidos de selectividad de H2 eran
similares. Una disminución de la temperatura aumentaba la relación
CO2/CO.
5. A medida que disminuye el tamaño de partícula de la fase activa de
níquel aumenta la selectividad de hidrógeno y la relación CO2/CO. A
menores tamaños se favorece las reacciones de reformado y oxidación

6. CONCLUSIONES
80
que dan lugar a la producción de hidrógeno. Los motivos principales de
la tendencia apreciada están actualmente en estudio y no se encontraron
referencias bibliográficas con qué compararlo.
6. La adición de un segundo lecho con catalizador WGS aumentó el
rendimiento global del proceso hacia la producción de hidrógeno. La
relación CO2/CO se incrementó considerablemente debido a la
conversión del monóxido de carbono.
7. A medida que disminuía la velocidad espacial del catalizador WGS, y
por tanto aumentaba el número de centros activos, se incrementaba la
relación H2/CO debido al desplazamiento del equilibrio hacia la
conversión de monóxido de carbono para producir hidrógeno.

7 RECOMENDACIONES

7. RECOMENDACIONES
82
A la vista de los resultados obtenidos, se proponen las siguientes recomendaciones
con el objeto de mejorar el rendimiento global del proceso de producción de hidrógeno:
1. Optimización de variables de reacción, tales como la relación
etanol/agua/oxígeno y la cantidad de agua incorporada a la entrada del lecho
adicional.
2. Estudio de otros métodos de síntesis del catalizador Ni-YSZ.
3. Ensayos catalíticos utilizando como soporte nanofibras de carbono, debido a
su elevada área superficial.
4. Síntesis de varios soportes de YSZ con distinta relación itria/zirconia, y su
posterior estudio del efecto sobre la distribución de productos de reacción.
5. Estudio del tamaño de partícula de YSZ en función del pH utilizado en su
síntesis, y su posterior relevancia en el proceso de reformado.
6. Análisis de microscopía electrónica de transmisión (TEM) para obtener
valores más precisos de diámetro de partícula.
7. Incorporación a los soportes de agentes dopantes con objeto de aumentar la
actividad del catalizador y la reducción de la cantidad de carbono
depositado. Se propone la adición de elementos lantánidos y alcalinotérreos
que proporcionen basicidad al soporte (tales como Ce, Mg o Ca), que

7. RECOMENDACIONES
83
pueden dar lugar a una neutralización de los centros de acidez para impedir
la deshidratación de etanol a otros compuestos orgánicos precursores de la
formación de coque.
8. Utilización de un reactor de membrana que realice la separación simultánea
del hidrógeno a medida que se va generando, con el fin de desplazar el
equilibrio de las reacciones hacia la formación del mismo.

8 ANEXO

8.1 ÍNDICE DE FIGURAS
Figura 2.1. Economía del Hidrógeno ............................................................................. 14
Figura 2.2. Producción de Hidrógeno a partir de diversas fuentes. ............................. 15
Figura 2.3. Producción de Hidrógeno puro. .................................................................. 17
Figura 2.4. Funcionamiento de una planta solar. .......................................................... 22
Figura 2.5. Funcionamiento del almacenamiento de energía eólica. ............................ 24
Figura 2.6. Reactor de alta temperatura y unidad de producción de hidrógeno........... 25
Figura 2.7.Distribución del consumo actual de Hidrógeno por aplicación. ................. 29
Figura 2.8. Esquema de funcionamiento de una pila de combustible de Hidrógeno-
Oxígeno. .......................................................................................................................... 33
Figura 4.1. Imagen del rotavapor. ................................................................................. 42

8. ANEXOS
86
Figura 4.2. Imagen de la mufla programable. ............................................................... 43
Figura 4.3. Imagen del espectrofotómetro. .................................................................... 45
Figura 4.4. Imagen del difractómetro. ........................................................................... 45
Figura 4.5. Imagen del modelo AUTOCHEM HP. ........................................................ 48
Figura 4.6. Imagen del equipo MICROMETRIC ASAP 2010. ....................................... 49
Figura 4.7. Esquema de la instalación experimental en ensayos con un lecho. ............ 51
Figura 4.8. Esquema de la instalación experimental en ensayos con doble lecho. ....... 51
Figura 4.9. Imagen del sistema de reacción. ................................................................. 53
Figura 4.10. Imagen del cromatógrafo HEWLETT PACKARD MODELO 5890. ......... 54
Figura 4.11. Imagen del espectrómetro de masas en un cromatógrafo SHIMADZU GC-
17A. ................................................................................................................................. 54
Figura 5.1. Difractograma del soporte YSZ. .................................................................. 59
Figura 5.2. Isoterma de adsorción-desorción y distribución de tamaño de poro del
soporte YSZ. .................................................................................................................... 60
Figura 5.3. Difractograma del catalizador YSZ-10%Ni. ............................................... 62
Figura 5.4. Difractograma del catalizador YSZ-15%Ni. ............................................... 62
Figura 5.5. Difractograma del catalizador YSZ-23%Ni. ............................................... 63
Figura 5.6. Digractograma del catalizador comercial WGS. ........................................ 64
Figura 5.7. Isoterma de adsorción-desorción y distribución de tamaño de poro del
catalizador YSZ-10%Ni. ................................................................................................. 65
Figura 5.8. Isoterma de adsorción-desorción y distribución de tamaño de poro del
catalizador YSZ-15%Ni. ................................................................................................. 65

8. ANEXOS
87
Figura 5.9. Isoterma de adsorción-desorción y distribución de tamaño de poro del
catalizador YSZ-23%Ni. ................................................................................................. 66
Figura 5.10. Ensayo de reducción térmica programada de los catalizadores 10, 15 y
23% Ni-YSZ. ................................................................................................................... 67
Figura 5.11. Ensayo de reducción térmica programada del catalizador comercial WGS.
........................................................................................................................................ 68
Figura 5.12. Relación CO2/CO (efecto de la temperatura). .......................................... 70
Figura 5.13. Relación CO2/CO (efecto del tamaño de partículas de níquel). ............... 72
Figura 5.14. Selectividad de hidrógeno en uno y dos lechos (efecto del tamaño de
partículas de níquel). ...................................................................................................... 74
Figura 5.15. Relación H2/CO en uno y dos lechos (efecto del tamaño de partículas de
níquel). ............................................................................................................................ 74
Figura 5.16. Relación CO2/CO en uno y dos lechos (efecto del tamaño de partículas de
níquel). ............................................................................................................................ 75
Figura 5.17. Comparación de la selectividad de gases (efecto de la velocidad espacial).
........................................................................................................................................ 76
Figura 5.18. Relación CO2/CO (efecto de la velocidad espacial). ................................ 77
8.2 ÍNDICE DE TABLAS
Tabla 2.1. Comparativa de los procesos de producción de hidrógeno. ......................... 26
Tabla 5.1. Propiedades texturales de los soportes. ........................................................ 60
Tabla 5.2. Contenido metálico de los catalizadores. ..................................................... 61
Tabla 5.3. Tamaño de partícula en función de la carga metálica ................................. 63

8. ANEXOS
88
Tabla 5.4. Propiedades texturales de los catalizadores. ................................................ 66
Tabla 5.5. Temperaturas máximas de reducción. .......................................................... 68
Tabla 5.6. Selectividad [%] de productos de reacción. ................................................. 69
Tabla 5.7. Conversión del etanol. .................................................................................. 70
Tabla 5.8. Selectividad [%] de los productos de reacción (efecto del tamaño de
partículas de níquel). ...................................................................................................... 71
Tabla 5.9. Conversión de etanol (efecto del tamaño de partículas de níquel). .............. 72
Tabla 5.10.Selectividad [%] de productos de reacción WGS (5000h-1
). ....................... 73
Tabla 5.11. Selectividad [%] de productos de reacción (efecto de la velocidad
espacial). ........................................................................................................................ 75

9 BIBLIOGRAFIA

9. BIBLIOGRAFÍA
90
i BP (2008) Statistical Review of World Energy, 2008, BP p.l.c. London.
ii J. Rifkin La economía del hidrógeno: La creación de la red energética mundial y la
redistribución del poder en la tierra, Barcelona, 2007.
iii Secretaría General de la Energía. La energía en España 2005. Ministerio de Industría,
Turismo y Comercio, Madrid, 2006.
iv European Commission (EU). Green Paper: A European Strategy for Sustainable,
Competitive and Secure Energy. COM, 105 final, 2006.
v European Commission (EU). Green Paper: Towards a European strategy for the
security of energy supply. COM, 769 final, 2000. Office for Official Publications of the
European Communities, Luxemburgo, 2001.
vi J.J. Brey, R. Brey, A.F. Carazo, I. Contreras, A.G. Hernández-Díaz, V. Gallardo.
Designing a gradual transition to a hydrogen economy in Spain. Journal of Power
Sources 159 (2006) 1231-1240.

9. BIBLIOGRAFÍA
91
vii
Intergovernmental Panel on Climate Change. Third Assessment Report: Climate
Change2001. Cambridge University Press, New York, 2001.
viii M. Momirlan, T.N.Veziroglu. The properties of hydrogen as fuel tomorrow in
sustainable energy system for a cleaner planet. International Journal of Hydrogen
Energy 30 (2005) 795-802.
ix European Commission (EU). White Paper: European transport policy for 2010: time
to decide. COM, 370 final, 2001. Office for Official Publications of the European
Communities, Luxemburgo, 2002.
x BOE núm. 33, Martes 8 febrero 2005, p. 4131.
xi BOE núm. 216, Martes 7 septiembre 2004, p. 30616.
xii Instituto para la Diversificación y el Ahorro de la Energía (IDAE). Estrategía de
Ahorro y Eficiencia Energética 2004-2012: Plan de Acción 2005-2007. Ministerio de
Industria, Turismo y comercio, 2005.
xiii Instituto para la Diversificación y el Ahorro de la Energía (IDAE). Plan de energías
renovables en España 2005-2010. Ministerio de Industria, Turismo y Comercio, 2005.
xiv Séptimo Programa Marco (2007-2013): Construir la Europa del conocimiento.
http://europa.eu/scadplus/leg/es/lvb/i23022.htm
xv Sexto Programa Marco (2000-2006): Desarrollo sostenible, cambio global y
ecosistemas. http://europa.eu/scadplus/leg/es/lvb/i23018.htm.
xvi A. Haryanto, S. Fernando, N. Murali, S. Adhikari. Current status of hydrogen
production techniques by steam reforming of ethanol: A review. Energy& Fuels 19
(2005) 2098-2106.
xvii S.Z. Baykara. Hydrogen as fuel: a critical technology?. International Journal of
Hydrogen Energy 30 (2005) 545-553.

9. BIBLIOGRAFÍA
92
xviii
M. L. Ghirardi, S. Kosourov y M. Seibert. Proceedings of the 2001 DOE Hydrogen
Program Review (2001).
xix B.C.R. Ewan, R.W.K. Allen. A figure of merit assessment of the routes to hydrogen.
International Journal of Hydrogen Energy 30 (2005) 809-819.
xx L.F. Brown. A comparative study of fuels for on-board hydrogen production for fuel-
cell-powered automobiles. International Journal of Hydrogen Energy 26 (2001) 381-
397.
xxi V. Klouz, V. Fierro, P. Denton, H. Katz, J.P. Lisse, S. Bouvot-Mauduit, C.
Mirodatos. Ethanol reforming for hydrogen production in a hybrid electric vehicle:
process optimisation. Journal of Power Sources 105 (2002) 26-34.
xxii M. Benito, J.L. Sanz, R. Isabel, R. Padilla, R. Arjona, L. Daza. Bio-ethanol steam
reforming: Insights on the mechanism for hydrogen production. Journal of power
Sources 151 (2005) 11-17.
xxiii Y. Yang, J. Ma, F. Wu. Production of hydrogen by steam reforming of ethanol over
a Ni/ZnO catalyst. International Journal of Hydrogen Energy31 (2006) 877-882.
xxiv A.N. Fatsikostas, X.E. Verykios. Reaction Network of Steam Reforming of Ethanol
over Ni-Based Catalysts. Journal of Catalysis 225 (2004) 439-452.
xxv E. Fakioglu, Y. Yürüm y T.N. Veziroglu. International Journal of Hydrogen Energy,
29 (2004) 1371-1376.
xxvi J. Pyle, R. Healy, Cortez R. Home Power 39, 32-38, (1994).
xxvii P. A. Lehman, C. E. Chamberlin, G. Pauletto, y M. A. Rocheleau. Proceedings del
10th
World Hydrogen Energy Conference Cocoa Beach, Florida (1994).
xxviii R. J. Friedland y A. J. Speranza. Proceedings of the2002 U.S. DOE Hydrogen
Program Review (2002).

9. BIBLIOGRAFÍA
93
xxix
R. Bocci. Workshop Environment and sustainable development: technical scientific
fundamentals for a hydrogen-based economy. Sesto Fiorentino (2003).
xxx Schatz Energy Research Center (2000).
http://www.schatzlab.org/spanish/solarh2cycle.html.
xxxi Parque eólico experimental sotavento (2007). http://sotaventogalicia.com/area-
tecnica/py-produccion-hidrogeno.php.
xxxii M. Ogawa, R. Hino, S.Shiozawa. Future Plan on Enviromentally Friendly
Hydrogen Production by Nuclear Energy (2002).
xxxiii . Dunn. International Journal of Hydrogen Energy, 27 (2002) 235-264.
xxxiv H. Cheng, Q. Yang, y C. Liu. Carbon, 39 (2001), 1447-1454.
xxxv S. Hynek, W. Fuller, y J. Bentley. International Journal of Hydrogen Energy, 22
(1997), 601-610.
xxxvi L. Zhou. Renewable and Sustainaible Energy Reviews, 9 (2005) 395-408.
xxxvii A. Züttel. MaterialsToday, September (2003) 24-33.
xxxviii A. Züttel. Naturwissenschaften, 91 (2004) 157-172.
xxxix .G. Tibbetts, G.P. Meisner y C.H. Olk. Carbon, 39(2001) 2291-2301.
xl M. Conte, P.P. Prosini y S. Passerini. Materials Science and Engineering B, 108
(2004) 2-8.
xli M.A. Laborde, M.C. Abello, P. Aguirre, N. Amadeo, J. Bussi, H. Corti, E González
Suarez, M.A. Gutiérrez Ortiz, V. Kajarov, A. Rodrígues. Produc ción y purificación de
hidrógeno a partir de bioetanol y su aplicación en pilas de combustible. CYTED Ciencia
y Tecnología para el Desarrollo Argentina, 2006.
xlii J.R. Rostrup-Nielsen, T. Rostrup-Nielsen. Large-scale hydrogen production
CATTECH6 (2002) 150-159.

9. BIBLIOGRAFÍA
94
xliii
Dra.C. Maria Teresa Hernández Nodarse, Tendencias actuales de la producción de
bioetanol.
xliv JohanssonT.B.: Kelly. H.: Reddy,AKN.; Williams, R.H. Renewables for fuels and
electricity, UNCED (June1992).
xlv S. Brunauer, P.H. Emmett, E. Teller. Adsorption of gases in multimolecular layers.
Journal of the American Chemical Society 60 (1938) 309-319.
xlviF. Rouquerol, J. Rouquerol, K. S. W. Sing. Handbook of Porous Materials. Wiley-
VCH, Weinheim, 2002, pp. 250.
xlviiE.P. Barret, L.G. Joyner, P.P. Halenda. The determination of pore volume and area
distributions in porous substances: I. Computations from nitrogen isotherms. Journal of
the American Chemical Society 73 (1951) 373-380.
xlviii X.S. Zhao, G. Q. (Max) Lu, G.J. Millar. Advances in mesoporousmolecular sieve
MCM-41. Industrial Engineering Chemical Resource35 (1996) 2075-2090.
xlix Ryoji Takahashi, Satoshi Sato, Satoshi Tomiyama, Tomoya Ohashi, Norifumi
Nakamura. Pore structure coltrol in Ni/SiO2
catalyst with both macropores and
mesopores 98 (2007) 107-114.
l Ataullah Khan, Panagiotis G. Smirniotis. Relationship between temperature-
programmed reduction profile and activity of modified ferrite-based catalysts for WGS
reaction. Journal of Molecular Catalysis A: Chemical 280 (2008) 43–5.
liP. Kim, Y. Kim, H. Kim, Y. K. Song, J. Yi. Appl. Catal. A 272 (2004) 157.
liiM. A. Tsurov, P. V. Afanasiev, V. V. Lunin. Appl. Catal. A 105 (1993) 205.
liii P. Kim, Y. Kim, C. Kim, H. Kim, Y. Park, J. H. Lee, I. K. Song, J. Yi. Catal. Lett. 89
(2003) 185.

9. BIBLIOGRAFÍA
95
liv
Nawadee Srisiriwat, Supaporn Therdthianwong, Apichai Therdthianwong. Oxidative
steam reforming of ethanol over Ni/Al2O3 catalysts promoted by CeO2, ZrO2 and
CeO2–ZrO2. international journal of hydrogen energy 34 (2009) 2224 – 2234.
lv M. Maron˜o, J.M. Sa´nchez, E. Ruiz. Hydrogen-rich gas production from oxygen
pressurized gasification of biomass using a Fe–Cr Water Gas Shift catalyst.
International journal of hydrogen energy 35 (2010) 37 – 45.


















