
Sindrome de guillain barre stroll
35
NEUROPATIAS SÍNDROME DE GUILLAN BARRE
-
Upload
eddynoy-velasquez -
Category
Health & Medicine
-
view
60 -
download
1
Transcript of Sindrome de guillain barre stroll

NEUROPATIASSÍNDROME DE
GUILLAN BARRE

DEFINICIONESNeuropatía periférica para referirsea aquellos trastornos de los nervios periféricos.Polineuropatía procesos de instauración gradual, que
afectan a múltiples troncos nerviosos y que se caracterizan por ser simétricos y generalizados, con afectación preferentemente distal.
Mononeuritis múltiple es una afectación simultánea o consecutiva de troncos nerviosos no contiguos, con evolución de días o años.
Mononeuropatías son afectaciones focales de un únicotronco nervioso.

CLASIFICACIONES



CLASIFICACIÓN GENERAL

SINDROME DEGUILLAN-BARRE

GENERALIDADES
El síndrome de Guillain Barré es una polineuropatía desmielinizante inflamatoria aguda de origen desconocido.
Su fisiopatología no está completamente aclarada y se señala que un organismo infeccioso induce una respuesta inmunológica, de origen tanto humoral como celular, la que produce una reacción cruzada contra la vaina de mielina de los nervios periféricos que causa su destrucción.

Definición
Es una poliradiculoneuropatia aguda marcada por parálisis flácida arreflexica; cuyas bases se consideran autoinmunes.
Actualmente se considera casi un síndrome con varios subtipos.
Journal Child of Neurology. Facultad de Medicina Universidad de Pensylvania

HISTORIAAunque la parálisis ascendente aguda ha sido reconocida por siglos,
una descripción aceptada fue ofrecida por Osler en 1882.
En 1916, Guillain, Barré y Strohl publicaron en París los elementos clínicos y los hallazgos del líquido cefalorraquídeo tal y como se conocen actualmente.
En 1949, Haymaker y Kernohen estudiaron material anatómico de soldados en la Segunda Guerra Mundial y consideraron el trastorno como una desmielinización.

En 1969, Asbury y otros reportaron los signos clínicos y los resultados de las autopsias de 19 pacientes fallecidos del síndrome, que fueron estudiados en vida y a los cuales se les realizó una extensa disección nerviosa, ya para 1978 propusieron criterios diagnósticos que son aceptados actualmente.
En la década de los 80, los trabajos de Osteman4 demostraron los efectos beneficiosos de la plasmaféresis, los que constituyeron junto al uso de las inmunoglobulinas, los 2 grandes avances en el tratamiento de la enfermedad.
Cont….
EpidemiologiaLa incidencia de SGB varió de 1,2/100.000 a 1,6/100.000 en los últimos
estudios europeos.
La incidencia aumenta con la edad pero en algunos estudios es bimodal, con un pico menor en los adultos jóvenes; es un poco más común en los hombres.
Se han descrito 12 casos de SGB familiar pero sin una relación firme con el HLA; sin embargo, un estudio halló una relación con un polimorfismo CD1.
La recurrencia del SGB es rara, y los pacientes con enfermedad crónica semejante a un SGB pero que tarda más de 4 semanas para alcanzar el nadir es clasificada como PrDI crónica (PrDIC). Estos pacientes tienen un comportamiento de la enfermedad diferente y suelen responder a los esteroides.

Patogenia


Inmunobiologia.
Anticuerpos antigangliosidos Mimetismo molecular y reactividad
cruzada Activación del complemento Factores del hospedador.

Anticuerpos antigangliosidos.

Procesos que anteceden al Síndrome de Guillain-Barré
1. Infecciones virales: Citomegalovirus, virusde Ebstein-Barr, herpes virus, VIH y otros menos frecuentes (hepatitis B, A y C, Sincitial respiratorio, Echovirus, Coxsackie, influenza A y B, adenovirus, parvovirus B19, sarampión, rubéola).
2. Infecciones bacterianas: Por Campylobacter jejuni, Mycoplasma pneumoniae y otras menos frecuentes (Salmonella typhi, Listeria, Brucella, Clamydia, Leptospira, Toxoplasma, Plasmodium del paludismo y Mycobacterium tuberculosis)
3. Cirugía4. Drogas: Zimelidina, sales de oro, D-penicilamina5. Enfermedades malignas: Linfoma de Hodgkin6. Vacunas: Antirrabia, antiinfluenza, anticolérica, antisarampión,
antirrubéola, antihepatitis B, antipoliomielitis7. Otros antecedentes: Alcoholismo con alteraciones hepáticas,
embarazo, puerperio, hipertiroidismo, insuficiencia adrenal

Cuadro clínicoLos síntomas iniciales consisten en sensación de “adormecimiento” y
“alfile-razos” en los dedos de los pies y en las manos, y en ocasiones por dolor en la región lumbar baja o en las piernas, seguido de debilidad muscular que suele iniciarse en los miembros inferiores para después afectar otros territorios.
Esta debilidad es a veces progresiva y puede afectar sucesivamente piernas, brazos, músculos respiratorios y pares craneales, todo lo cual configura el cuadro clínico de parálisis ascendente de Landry.


La afectación de pares craneales ocurre en el 25 % de los casos, siendo la paresia facial bilateral la más característica, aunque también pueden ocurrir debilidad en los músculos de la deglución, fonación y masticación.
Los signos de disfunción autonómica están presentes en el 30 al 50 %, entre ellos se encuentran:
Arritmias (bradicardia, taquicardia paroxística así como asistolia). Hipotensión ortostática. Hipertensión arterial transitoria o permanente. Íleo paralítico y disfunción vesical. Anormalidades de la sudación.

Las manifestaciones clínicas de debilidad de la musculatura respiratoria incluyen:
Taquipnea mayor de 35 por minutos. Reducción del volumen tidal en menos de 4mL/kg. Movimiento paradójico abdominal (movimiento hacia dentro
durante la inspiración). Alternancia respiratoria (alternativa entre movimientos
predominantemente abdominales y los de la caja torácica durante la inspiración).
Kuwabara S. Guillain-Barre síndrome: epidemiology, pathophysiology and management. Drugs. 2004:64(6):597-610.

VARIANTES CLÍNICAS Polineuropatía Desmielinizante Inflamatoria Aguda (AIDP): afecta más
a los adultos que a los niños. El 90% de los casos ocurre en occidente. Los pacientes presentan anticuerpos anti GM1, y la recuperación es rápida. Es desmielinizante. Primero ataca la superficie de las células de Schwann, provocando daño en la mielina. Hay activación de macrófagos e infiltrado linfocítico.
Neuropatía Axonal Motora Aguda (AMAN): afecta a jóvenes y adultos. Es prevalente en china y México, puede ser estacional. Rápida recuperación. Presentan anticuerpos anti GD1a. Es axonal. Primero ataca los nódulos de Ranvier de los nervios motores. Hay activación de macrófagos, pocos linfocitos y es frecuente la presencia de macrófagos periaxonales. Hay daño axonal extenso.

Neuropatía Axonal Motora y Sensorial Aguda (AMSAN): afecta preferentemente a adultos. Es poco común. La recuperación es lenta. Está relacionada con AMAN. Es axonal. Es similar a AMAN pero también afecta nervios sensoriales y raíces de nervios. El daño axonal es frecuentemente severo.
Síndrome de Miller Fisher (MFS): afecta a adultos y niños. Es poco frecuente (aproximadamente 5% de los casos de SGB). Causa oftalmoplejía, generalmente con parálisis pupilar; ataxia y arreflexia. Los pacientes presentan anticuerpos anti GQ1b (90%). Es desmielinizante. No se conocen muchos casos. Es similar a AIDP.
Hahn A. F. Guillain-Barré Syndrome. Lancet 1998; 352: 635-641.

DIAGNOSTICO

1. Hallazgos necesarios para hacer el diagnóstico
- Debilidad progresiva en varias extremidades- Arreflexia
Cont….
3. Hallazgos que hacen el diagnóstico dudoso- Existencia de un nivel sensorial- Marcada asimetría de síntomas y signos- Disfunción severa y persistente de vejiga e intestino- Más de 50 células/ mm3 en LCR
4. Hallazgos que excluyen el diagnóstico- Diagnóstico de botulismo, miastenia, poliomielitis o neuropatía tóxica- Metabolismo alterado de las porfirinas- Difteria reciente- Síndrome sensorial puro sin fatiga
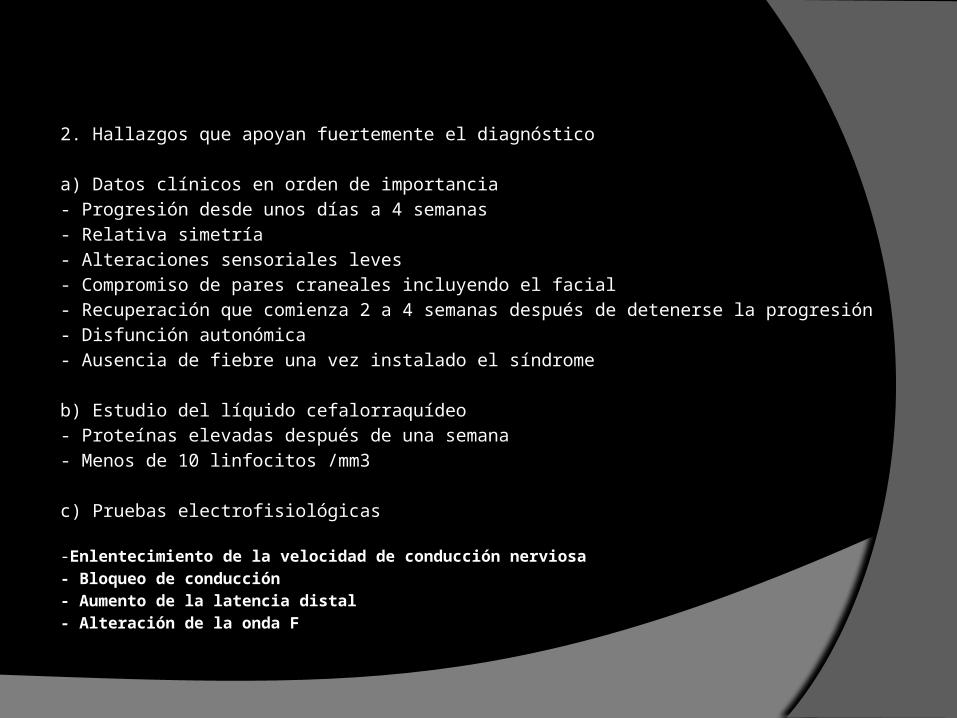
2. Hallazgos que apoyan fuertemente el diagnóstico
a) Datos clínicos en orden de importancia- Progresión desde unos días a 4 semanas- Relativa simetría- Alteraciones sensoriales leves- Compromiso de pares craneales incluyendo el facial- Recuperación que comienza 2 a 4 semanas después de detenerse la progresión- Disfunción autonómica- Ausencia de fiebre una vez instalado el síndrome
b) Estudio del líquido cefalorraquídeo- Proteínas elevadas después de una semana- Menos de 10 linfocitos /mm3
c) Pruebas electrofisiológicas -Enlentecimiento de la velocidad de conducción nerviosa- Bloqueo de conducción- Aumento de la latencia distal- Alteración de la onda F

Estudios electrofisiológicosSe observa bloqueo de conducción parcial, con algún grado de dispersión temporal
anormal y marcada lentitud de la conducción en el segmento bloqueado.
Latencia distal normal.
La respuesta sensitiva tiene amplitud disminuida o está ausente en los segmentos distales del nervio, que tiene bloqueada la conducción proximal.
El bloqueo persiste por largos períodos de tiempo y afecta solo a axones motores, sin bloquear la conducción sensitiva.
La velocidad de conducción está enlentecida en el segmento bloqueado.
Los potenciales evocados somatosensoriales son normales.
La electromiografía muestra signos de denervación, fasciculaciones o mioquimias, o ambas, en los músculos afectados.
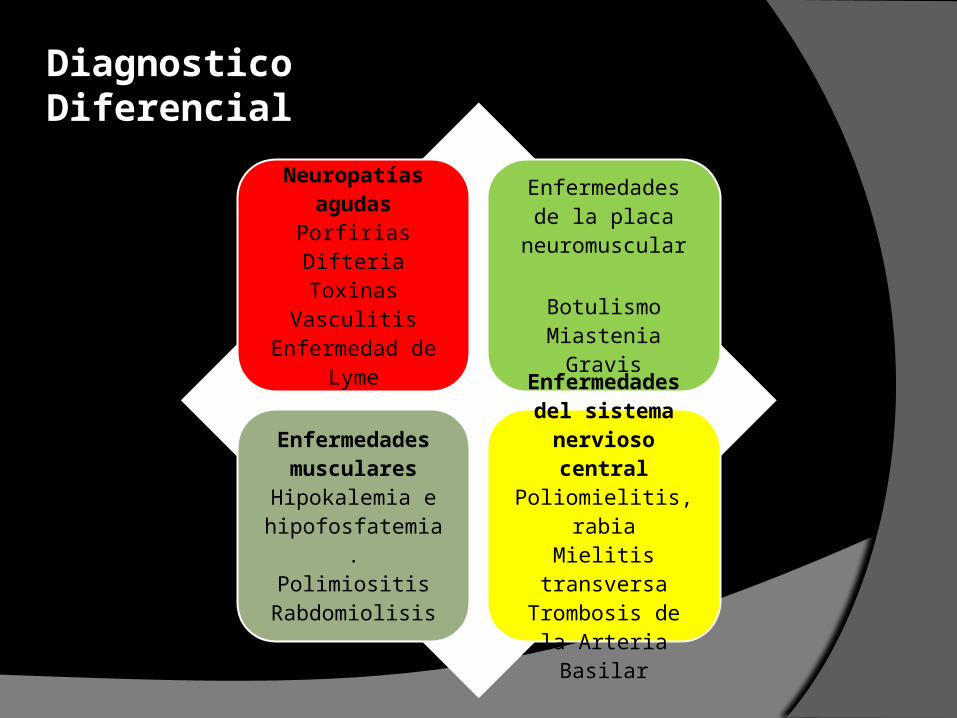
Neuropatías agudasPorfiriasDifteriaToxinas
VasculitisEnfermedad de
Lyme
Enfermedades de la placa
neuromuscular
BotulismoMiastenia Gravis
Enfermedades musculares
Hipokalemia e hipofosfatemia.
PolimiositisRabdomiolisis
Enfermedades del sistema nervioso
centralPoliomielitis, rabiaMielitis transversaTrombosis de la Arteria Basilar
Diagnostico Diferencial


Trastornos de nervios periféricos, con síntomas predominantemente motores:
Síndrome de Guillain- Barré, polineuropatía subaguda y crónica, con hiperproteinorraquia; polineuropatía crónica recurrente, porfiria intermitente aguda, neuropatía saturnina, neuropatía diftérica, neuropatías hipertróficas (enfermedad de Charcot - Marie- Tooth y enfermedad de Dejerine-Sottas), neuropatía motora Multifocal.

Trastornos de nervios periféricos con síntomas predominantemente sensitivos:
Lepra, déficit de vitamina B1, medicamentosas, por arsénico, en el desarrollo de amiloidosis, en la evolución del mieloma múltiple, diabetes sacarina, polineuropatía isquémica, polineuropatía diabética, polineuropatía urémica

Trastornos de la célula del ganglio posterior:
Neuropatía radicular sensitivo-hereditaria, neuropatía sensitiva congénita, neuropatía sensitiva carcinomatosa, insensibilidad congénita al dolor, degeneración progresiva de células ganglionares de la raíz posterior, sin carcinoma; ataxia de Friederich, herpes zoster.

Tratamiento Plasmaféresis. Régimen usual: 5 PE en un periodo
de 2 semanas, realizando recambios totales de volumen de plasma.
Inmunoglobulina Intravenosa. Dosis: 0.4 g/kg durante 5 días continuos.
Esteroides (controvertido). Dosis: 500 mg durante 5 días.
Moduladores de la respuesta inmunológica.Micofenolato mofetil.Anticuerpos monoclonales que intervienen con la activación del complemento

Evolución y pronosticoLa enfermedad evoluciona en 3 fases, denominadas: de progresión,
estabilización y regresión, que suele completarse en 3 a 6 meses.
El 80 % de los pacientes se recuperan completamente o con déficit pequeños. Entre el 10 y el 15 % quedarán con secuelas permanentes; el resto morirá a pesar de los cuidados intensivos.
Las causas de muerte incluyen: distrés respiratorio agudo, neumonía nosocomial, broncoaspiración, paro cardíaco inexplicable y tromboembolismo pulmonar.

Los factores asociados con un mal pronóstico son: Edad mayor de 60 a. Progresión rápida de la enfermedad. Extensión y severidad del daño axonal (amplitud
motora distal media menor del20 % de lo normal). Enfermedad cardiorrespiratoria preexistente. Tratamiento tardío.


















