
SíNDROME DE ALB RIGHT P. PIULACHS, E. VENDRELL-TORNÉ, E ...
20
Sesión del día 12 de enero de 1972 SíNDROME DE ALBRIGHT P. PIULACHS, E. VENDRELL-TORNÉ, E. LLAURADÓ-MIRET El síndrome de Albright es un cuadro caracterizado por la siguien- te tríada: 1) lesiones óseas, casi siempre unilaterales, con el aspecto de una osteítis fibrosaj 2) pigmentación cutánea en forma de gran- des manchas de color café con leche, generalmente predominando en el mismo lado en que existen las lesiones óseas; 3) disfunción endo- cril?a traducida por trastornos del crecimiento y, sobre todo, por pu- bertad precoz en las mujeres. Fue descrita por AL.BRlGHT, BuTLER, HAMPTON y SMITH, 2 quienes en 1936 publicaron ci.nco casos. Anteriormente, WEIL 49 (1922) había comunicado un caso de una niña de nueve años que tenía hemorragias vaginales periódicas desde los dieciocho meses y había sufrido ocho fracturas espontáneas. La piel estaba anormalmente pigmentada y la edad ósea avanzada. Los huesos presentaban una imagen radiológica que fue etiquetada de osteopsa- tirosis. En 1938, LICHTENSTEIN a.¡ demostró que lesiones óseas similares a las vistas en el síndrome de Albdght pueden presentarse como fenó- meno aislado, y dio a esta anomalía ósea el nombre de «displasia fibro- sa poliostótica». Más tarde se demostró que las alteraciones óseas pue- den quedar limitadas a un solo hueso, por lo cual }AFFE y LrcHTENS- TEIN 35 (1942) cambiaron aquel nombre por el de «displasia fibrosa de los huesos». La etiopatogenia del síndrome de Albrigth es desconocida; en la actualidad se considera al proceso, como un trastorno embriológico de origen neural que probablemente asentaría en el hipotálamo, lo que explicaría la asociación de trastornos endocrinos. No hay que confundir este síndrome de Albright con el síndrome de BuTLER-ALBRIGHT, que es una osteodistrofia renal por nefropatía tu- bular crónica, afección en la que existe un déficit de la amoniogénesis,
Transcript of SíNDROME DE ALB RIGHT P. PIULACHS, E. VENDRELL-TORNÉ, E ...

Sesión del día 12 de enero de 1972
SíNDROME DE ALBRIGHT
P. PIULACHS, E. VENDRELL-TORNÉ, E . LLAURADÓ-MIRET
El síndrome de Albright es un cuadro caracterizado por la siguiente tríada: 1) lesiones óseas, casi siempre unilaterales, con el aspecto de una osteítis fibrosaj 2) pigmentación cutánea en forma de grandes manchas de color café con leche, generalmente predominando en el mismo lado en que existen las lesiones óseas; 3) disfunción endocril?a traducida por trastornos del crecimiento y, sobre todo, por pubertad precoz en las mujeres.
Fue descrita por AL.BRlGHT, BuTLER, HAMPTON y SMITH,2 quienes en 1936 publicaron ci.nco casos.
Anteriormente, WEIL 49 (1922) había comunicado un caso de una niña de nueve años que tenía hemorragias vaginales periódicas desde los dieciocho meses y había sufrido ocho fracturas espontáneas. La piel estaba anormalmente pigmentada y la edad ósea avanzada. Los huesos presentaban una imagen radiológica que fue etiquetada de osteopsatirosis.
En 1938, LICHTENSTEIN a.¡ demostró que lesiones óseas similares a las vistas en el síndrome de Albdght pueden presentarse como fenómeno aislado, y dio a esta anomalía ósea el nombre de «displasia fibrosa poliostótica». Más tarde se demostró que las alteraciones óseas pueden quedar limitadas a un solo hueso, por lo cual }AFFE y LrcHTENSTEIN 35 (1942) cambiaron aquel nombre por el de «displasia fibrosa de los huesos».
La etiopatogenia del síndrome de Albrigth es desconocida; en la actualidad se considera al proceso, como un trastorno embriológico de origen neural que probablemente asentaría en el hipotálamo, lo que explicaría la asociación de trastornos endocrinos.
No hay que confundir este síndrome de Albright con el síndrome de BuTLER-ALBRIGHT, que es una osteodistrofia renal por nefropatía tubular crónica, afección en la que existe un déficit de la amoniogénesis,

138 Al'IALES DE MEDICINA
por lo que el organismo tiene que echar mano de las bases de la sangre para la eliminación de los ácidos. Existe una eliminación excesiva por la orina, de sodio, potasio, magnesio y también de calcio. Esta hipercalciuria se acompaña de hipocalcemia y de hipofosforemia, alteraciones metabólicas que conducen, en el niño, a un raquitismo, y, en el adulto, a una osteomalacia.
FRECUENCIA.- En tanto que la displasia fibrosa es relativamente frecuente, el síndrome de Albright completo es raro. En una revisión de la literatura efectuada en 1949, PRITCHARD 42 econtró 256 casos de displasia fibrosa, de los cuales 181 eran poliostóticos. De éstos, 79 tenían pigmentación anormal de la piel, mientras que 37 de las 82 mujeres y 5 de los 83 varones, presentaban pubertad precoz.
En Escandinavia, solamente HERNBERG y EnGREN (1949) han ptl
blicado un síndrome de Albrigbt en una niña de 14 años, la cual, sin embargo, no tenía signos de precocidad sexual.
En 1968, MARTÍN MuÑoz 39 indica que el número de casos publicados sería aproximadamente de unos 125. La frecuencia del síndrome de Albright correspondería a la mitad de la observada en la enfermedad de }AFFE-LICHTENSTEIN.
CALDWELL y BRODERICK 12 en 194 7 publicaron el primer caso en la raza negra, en una niña de nueve años. En 1965, CAPLAN y FEINGOLD 13 describieron la primera observación en un negro de sexo masculino.
En 194 9, LARA, CA S TILLO y FRAi'lCO 31 publican un caso; no tienen noticia de que en España se haya publicado ninguno antes del suyo. Ulteriormente han sido publicados, en nuestro país, cinco nuevos casos: SÁNCIIEZ-LUCAS y CASTELLS 46 (1949), BERNÁLDEZ 9 (1958), RAVENTÓS, DARGALLO y TURON 44 (1959), PROFITOS 43 (1964) y MAR· TÍN MuÑoz 39 (1968).
Es bastante más frecuente en la mujer que en el varón. No suele haber historia familiar, pero no es infrecuente que se
descubran discromías en algún pariente cercano, casi siempre la madre.
Nosotros hemos tenido ocasión de observar el siguiente caso de síndrome de Albright que describimos a continuación:
J. V. C., enfermo de 24 años de edad. Ingresa en nuestro Servicio el 15-11-65.
Antecedentes fisiológicos: Nacido a término, de parto eutócico. Lactancia materna. Fonación y deambulación normales. Desarrollo intelectual y edad escolar normales.

PIULACHS Y COLS. SÍNDROME DE ALBRlGIIT 139
Antecedentes patológicos: A la edad de diez años, sarampión. Amigdalit is y gripes aisladas.
Hace nueve años, por caída fortuita estando en su domicilio, se produjo una fractura de la cadera izquierda, con intenso dolor, tumefacción e impotencia funcional completa. Al día siguiente fue trasladado al hospital. Fue tratado con tracción continua durante rres meses. Un mes más tarde regresó al trabajo.
Enfermedad actual: El enfermo precisa que hace unos cinco años que empieza a notar deformidades en la región dorsal y en la hemicara derecha.
Hace dos años, al principio de invierno, apareció dolor en la cadera izquierda, que aumentaba con la deambulación y que a veces se irradiaba hacia la cara anteroexterna del muslo. A los tres meses este dolor aparecía andando y en reposo, y sólo calmaba cuando el paciente dormía. Dice que un mes más tarde fue intervenido de la cadera izquierda.
Ftc. l.- Aspecro general del paciente. Abombamiento de la región frontoparietal derecha; escoliosis dorsal baja, de concavidad izquierda; acortamiento de la e.'(tremidad inferior izquierda. En la región supraclavicular derecha, brazo derecho, bemit6rax izquierdo y nalga derecha, manchas de café con leche con las características propias de las discromías del síndrome de Albright: localización troncular, variaciones de la intensidnd de la pigmen-
tación, contorno escarpado, terminación en la linea media.

140 ANALES DE MEDICINA
Frc. 2. - Abombamiento de la región frontal derecha, que nlcnnza el techo de la órbita,
lo cual provoca un descenso y pro[rusióo del globo ocular del mismo lado. Mancha de café con leche en la mejilla izquierda.
En el intervalo existente entre la apartcton de las molestias y la intcl"vención, el enfermo parece apreciar que la extremidad inferior izquierda ha
disminuido de longitud. Hace unos diez meses empieza un episodio de astenia; al cabo de un
mes aparecen molestias y dolor en la región dorsal, de preferencia por la
tarde. Un mes y medio después los dolores disminuyen y se trasladan a la región lumbosacra. Calman con el reposo.
Actualmente los dolores se acentúan con el paciente en posición erecta y disminuyen con la deambulación y el reposo.
Exploración: Por inspección se aprecia (figs. 1 y 2) marcada prominencia de la región frontoparietal derecha. Exoftalmos en el lado derecho. Escoliosis dorsal baja de concavidad izquierda (fig. 1 ). En la cara externa del muslo se aprecia una cicatriz operatoria de unos veinte centímetros. En la cara anterointerna de la pierna izquierda hay otra cicatriz de igual longitud.
Acortamiento de seis centímetros del miembro inferior izquierdo. Manchas pigmentarias (figs. 1 y 2) de color de café con leche, de bor
des irregulares, en la cara externa del cuello y hombro y en la cara antcroex-

PI ULAC HS Y COLS. SÍNDROME DE ALBRIGHT 141
FJG. 3.- AspccLo de las menos del paciente: dedos cortos. En la mano dc1·echn ~e ap¡·ccia engl'osomienLO del bo1·de cubita l del quin to metacarpiano, y de la primera fa lange del
se¡¡undo dedo )' segunda falange del tercero.
1-rG. -1.- Escoliosis dorsal media.

142 ANALES DE MEOICL'lA
na del brazo derecho; en la mejilla izquierda; en la cara anterior, lateral y posterior del hemitórax izquierdo; en la región glútea derecha y en la cara posterosuperior del muslo izquierdo. Hace unos doce años aparecieron manchas en el labio inferior.
Los dedos son cortos (fig. 3 ); los de la mano derecha aparecen más gruesos que los de la izquierda, sobre todo en la primera y segunda falanges.
1
FJG. 5.-Húmero de aspecto rad iológico totalmente superponible al de lA displasia fibroso: insuflación de la diáfisis, adelgazamiento de la cortical, imágenes geódicas en cuyo fondo ha desaparecido la trabecu~ación ósea normal, que es sustituida por una imagen de claridad homo·
génea.

PIULACIIS Y COLS. SÍNDROME DE ALBRlGHT
FtG. 6. - Radiograffa del antebrazo derecho. Se aprecia incurvaci6n de la diáfisis radial, con altérnaocia de zonas condensadas con zonas claras.
143
Hay abombamiento del quinto metacarpiano derecho y de la primera falange del segundo dedo y segunda falange del tercero.
Por palpación se aprecia la apófisis mastoides detecha muy desarrollada. Dolor a la percusión en las apófisis espinosas de la región lumbosacra. Dolor a la percusión en la zona de cicatriz de la pierna izquierda.
Examen radiológico: La radiografía del tórax (fig. 4) pone de manifiesto la escoliosis dorsal media. En el resto del esqueleto se aprecian imágenes po· liostóticas y poli tópicas de osteítis fibrosa:
En el húmero derecho (fig. 5) se observan lesiones típicas de displasia fibrosa: insuflación de la diáfisis, adelgazamiento de la cortical, imágenes geódicas en cuyo fondo ha desaparecido la trabeculación ósea normal, que es sustituida por una imagen de claridad homogénea. En la radiografía del antebrazo (fig. 6) se aprecia incurvación de la diáfisis radial, con alternancia

144 ANALES DE MEDICL'L\
de zonas condensadas y zonas claras. La radiografía de las manos (fig. 7) permite observar: en el lado derecho abombamiento del quinto metacarpiano y de la primera falange del segundo dedo; ambos ofrecen aspecto radiográfico parecido al del húmero. Lesiones geódicas en el tercero y cuarto metacarpianos y en las falanges primera y segunda del quinto dedo. En las primeras van imágenes geódicas muy parecidas a las que se aprecian en los casos de encondromas de la mano.
La diáfisis en ambos fémures (fig. 8) presenta lesiones parecidas a las del húmero, aunque menos acusadas. En la metáfisis de la tibia derecha (fig. 9) se aprecia una larga geoda de bordes policíclicos, cuyo contorno presenta una condensación ósea que la hace destacar muy claramente. En el calcáneo derecho (fig. 10) se observa una gran geoda subtalámica, cuyo interior ofrece aspecto trabecular. En el calcáneo izgLúerdo (fig. 11) se aprecia una voluminosa geofalange de los cuatro tútimos dedos ele la mano izquierda, sobre todo en las de los dedos tercero y cuarto, se observa, limitada por un neto reborde de condensación.
Examenes de laboratorio: Hematíes, 5.200.000 por milímetro cúbico; hemoglobina, 105 %; valor globular, 0,98; leucocitos, 5.500; en banda, 3 %; segmentados neutrófilos, 56%; eosinófilos, O%; basófilos, O%;
t
F1c. 7. - El quinto metacarpiano de la mano derecha presenta un aspecto radiográfico parecido al del húmero. En diversos metacarpianos y falanges se aprecian imágenes ge6-
dicas muy parecidas a las que se observan en los encondromas de esta región.

PIULACHS Y COLS. SÍNDROME DE At.BRIGHT
ftG. 8. - Diáfisis femorales, con lesiones parecidas a las del húmero, aunque menos acusadas.
145
linfocitos, 37 %; monocitos, 4 %. Erin:osedimentacíón : 1.a hora, 1 mm.; 2.• hora, 4 mm.; índice Katz, 1,50.
Uremia, 42 mg. %; glucemia, 73 mg. %; tiempo de coagulación, 31 minutos; tiempo de sangría, 4 minutos; fósforo en el suero, 4,1 mg. %; fósforo en la orina, 147 mg. %; calcemia, 9,6 mg. %; magnesiemia, 2,6 mg. %; fosfatasas, 13,5 u. Bodansky; calciuría, 3-lO mg. %.
Examen histológico (Dr. RuBIO). Se efectúa una biopsia ósea en el quinto metacarpiano derecho y otra en la tibia derecha. Biopsia de un fragmento de piel del cuello, que acabalga sobre el borde de una mancha de café con · leche.
La biopsia cutánea (fig. 14) demuestra intensa pigmentación melánica de la basal epidérmica y en los melanóforos dérmicos.
ANATOMÍA PATOLÓGICA.- El aspecto histológico de las lesiones óseas en el síndrome de Albright es el característico de la displasia fibrosa. Además de las células espumosas y las células gigantes multinucleadas identificables como fagocitos, muchos autores describen la existencia de células gigantes indistinguibles de los osteoclastos. En

146 At'IIALES DE :llEDICINA
FrG. 9. -En la metáfisis de la tibia derecha se aprecia una larga geoda de bordes policí
clicos cuyo contorno presenra una condensación ósea que la hace destacar claramente.
FrG. 10. - Radiograffa del pie derecho. Se aprecia en el calcáneo una gran geoda subta·
Jámica cuyo interior ofrece aspecto trabecular.
F!G. 11. - Radiografía de perfil del pie i7.quierdo. Se aprecia una geoda muy voluminosa
en el calcáneo; está limitada por un marcado reborde de condensación.
dos casos de DOCKERTY, GHORMI.EY, KENNEDY y PuGH 15 (1945) las
células gigantes con 50 a 60 núcleos eran tan numerosas gue el as
pecto histológico era el de un tumor de células gigantes.
PlULi\CITS Y COLS. SÍNDROME DE ALBRIGHT 147
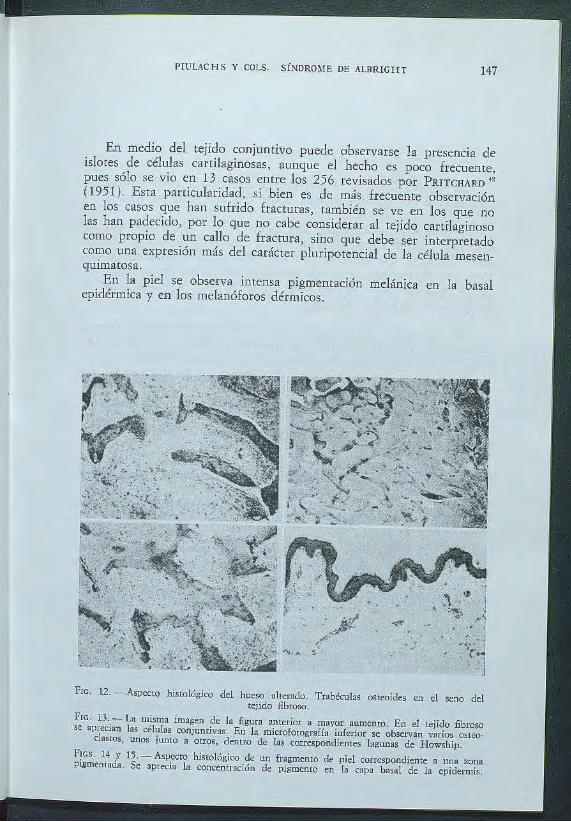
En medio del tejido conjuntivo puede observarse la presencia de islotes de células cartilaginosas, aunque el hecho es poco frecuente, pues sólo se vio en 13 casos entre los 256 revisados por PRITCIIARD 42
(1951 ). Esta particularidad, si bien es de más frecuente observación en los casos que han sufrido fracturas, también se ve en los que no las han padecido, por lo que no cabe considerar al tejido cartilaginoso como propio de un callo de fractura, sino que debe ser interpretado como una expresión más del carácter pluripotencial de la célula mesenquimatosa.
En la piel se observa intensa pigmentación melánica en la basal epidérmica y en los melanóforos dérmicos.
. . 1 -. '• " . \.
Frc. 12. -Aspecto histológico del hueso alterado. Trabéculas osteoides en el seno del teíido fibroso.
Ftc. 13.- La misma imagen de la figura anterior a mayor aumento. En el teíido fibroso se aprecian las células coníuntivas. En la microfotografía inferior se observan varios osteo·
clastos, unos íunto a otros, dentro de las correspondientes bgunas de Howship.
FIGS. 14 y 15. -Aspecto histológico de un fragmento de piel correspondiente a una zona pigmentada. Se aprecia la concentración de pigmemo en la capa basal de la epidermis.

148 ANALES DE M EiDlCINA
CLÍNICA.- A) LESIONES OSEAS
Las lesiones óseas de la enfermedad de Albright pueden presentar
clínicamente tres manifestaciones: dolor, fracturas y deformaciones.
I) Dolor. Es inconstante; hay enfermos que no lo presentan. En
los casos en que existe, se trata, en general, de dolor moderado, de
corta duración, que empeora con las malas condiciones atmosféricas.
Pero hay un pequeño grupo de enfermos que sufren dolores muy in
tensos, casi insoportables, de carácter terebrante, osteócopo, que se
localizan en los huesos afectos.
II) Fracturas. En general, es el accidente que, al exigir el examen
radiográfico, permite descubrir la enfermedad. Según LAYANI y Du
RUPT 32
( 1951) estas fracturas presentan los siguientes caracteres:
a) Asientan sobre los huesos displásicos, fragilizados por las nu
merosas zonas seudoquísticas que los pueblan.
b) Se producen con traumatismos mínimos; son hacturas espon
táneas. e) Son iterativas; de hecho constituyen la única gran complica
ción de la enfermedad. d) Frecuentemente son poco dolorosas, de forma que pueden in
cluso pasar inadvertidas y ser descubiertas al efectuar un examen radio
gráfico sistemático. e) Consolidan bien y rápidamente, siendo rarísimos los casos de
seudartrosis.
III) Deformaciones. Aunque pueden deformarse todos los huesos,
las deformaciones más importantes se producen en los huesos largos
de las extremidades inferiores, debido al peso que han de soportar.
El hueso que más frecuentemente se afecta de displasia y que con
mayor frecuencia se deforma es el fémur, que sufre una progresiva
incurvación, tanto en el cue1lo como en la diáfisis, adquiriendo la clá
sica forma en «cayado de pastor» o en «báculo episcopal».
Estas deformaciones son debidas a la blandura del hueso y a las
frecuentes fracturas del mismo. Conducen a un acortamiento más o
menos acusado de la extremidad. Existen también abombamientos localizados debido a las insuficien
cias óseas . Los engrosamientos más acentuados se presentan en el ma
cizo facial y en la bóveda craneal. Topografía de las lesiones óseas. Las modalidades topográficas de
las lesiones óseas de la displasia fibrosa pueden clasificarse en circo
grupos:

PlliL.-\CHS Y COLS. SÍ:"lDROME DE ALBRIGHT 149
1) Formas monostóticas. 2) Formas oligost6ticas. 3) Formas unilaterales, que pueden ser de tres tipos:
a) tipo monomético o axil de Kienbock, en el gue se afectan la parte externa de la escápula, el húmero, el radio y el primer metacarpiano o bien, la hemipelvis (excluido el sacro), el fémur, la tibia y el primer metatarsiano.
b) tipo bimélico que afecta al miembro superior y al inferior del mismo lado.
e) tipo hemiesquelético que corresponde a los casos de afectación bimélica a los que se suma la de la mitad correspondiente de la cara y cráneo homolaterales.
4) Formas diseminadas o bilaterales poliostóticas. 5) Formas cefálicas puras, hasta hace poco etiquetadas de leon
tiasis ósea o de enfermedad de Paget juvenil. En el síndrome de Albright no se observan formas monostóticas ni
oligostóticas.
B) PIGMENTACIONES CUTANEAS. ~lB En general las pigmentaciones cutáneas se desarrollan entre los
cuatro meses y los dos años de edad, pero se han observado también en las primeras semanas de la vida BERARDINELLI/ (1950).
BENEDICT, SZABO, FITZPATRICK y SINESI ' (1868) comparan los datos clínicos de las pigmentaciones cutáneas de 27 casos de síndrome de Albl"ight con los de 19 casos de neuwfibromatosis, y resumen los caracteres diferenciales entre ambos en el cuadro de la páginas siguiente.
Estos datos diferenciales, sin embargo, no son lo suficientemente constantes para permitir establecer el diagnóstico diferencial entre ambas afecciones. Los citados autores presentan ejemplos de neurofibromatosis en los que las manchas tenían las características propias del síndrome de Albright. Por ello creen gue, aparte la pigmentación pecosa de la axila, la cual consideran totalmente típica de la neurofibromatosis, no existen datos clínicos suficientemente seguros para el diagnóst ico difeJ"encial.
BENEDICT, SzABO, FITZPATRICK y SINESI/ (1968) han verificado un estudio microscópico de biopsias cutáneas en los 19 casos de neurofibromatosis y en 10 de los 27 casos de síndrome de Albright. Comprobaron que en 18 de los 19 casos de neurofibromatosis existían gránulos gigantes de pigmentos en las células malpighianas, en los melanocitos, o en ambos, tanto en la piel de zonas normales como en la piel de las zonas pigmentarias. Estos gránulos esféricos eran de tamaño variado; unos llenaban casi toda la célula, y otros eran muy pequeños,

150 ANALES DE ~IEDJClNA
Sindrome de Albright
.Patrón de distribución
Tienden a colocarse a rambos lados de la línea media. Suelen ser unilaterales y situadas en áreas determinadas: cuello, nalgas, tórax.
Número Pocas. de lesiones
Configu- Bordes escarpados, en cos-ración ta recortada (del Maine,
Costa Brava, Noruega).
Color Además de las manchas de «café con leche», hay otras de castaño algo más subido, e incluso de castaño oscuro. En el cuero cabelludo, los cabellos correspondientes al área de la lesión pueden ser más pigmentados que los vecinos.
Tamaño Desde pocos centímetros a grandes áreas.
Neurofibromatosis
Tienden a ser generalizadas y de distribución irregular. La pigmentación, en forma de pecas, de la axila, es característica de la neurofibroma tosis.
Muchas.
Bordes más suaves.
Tinte «café con leche».
Además de las manchas de café con leche, puede haber máculas en forma de pecas generalizadas.
en el límite de resolución del microscopio óptico (aproximadamente o,5 IJ.). Su naturaleza química no es aún conocida.
Entre los 10 casos de síndrome de Albríght, sólo en uno se hallaron estos gránulos.
Estas proporciones son estadísticamente significativas y permiten a BENEDICT, SZABO, FITZPATRICK y SrNESI
7 ( 1968) proponer la biopsia
cutánea, en busca de estos gránulos pigmentarios, como el método más seguro para diferenciar las manchas cutáneas del Albrigbt de las de la neurofibromatosis.

PIULACHS Y COLS. SÍNDROME DE ALBRIGHT 151
C) PUBERTAD PRECOZ Aproximadamente en la mitad de las mnas con síndrome de Al
brigbt la menarquia se produce en la edad de uno a cinco años, y en una tercera parte de los casos, entre los seis y diez años.
No obstante, se han observado hemorragias vaginales a la edad de cuatro meses (ALBRIGTH y colaboradores 2 1936); e incluso en el segundo día de la vida (HACKETT y CHRISTOPHERSON 25 1949).
La hemorragia persiste, en general, de dos a cuatro días y, en ocasiones, es abundante. Puede ser regular o irregular, apareciendo a intervalos de semanas o meses.
El desarrollo de las mamas y la aparición del vello público )' axilar suelen ser más tardíos que la menarquia, generalmente entre los cinco y los diez años de edad, pero se han presentado también a los dos años (McUNE,36 McUNE y BRUCH,37 1937). ALBRIGHT y colaboradores 2
( 1936) han observado hipertrofia de los genitales externos a los seis meses de edad.
Se ha publicado algún caso de pigmentaciones y precocidad sexual sin acompañarse de lesiones óseas, asociación que más tarde ha sido denominada displasia fibrosa poliostótica sin displasia fibrosa.
Además de la pubertad precoz se han descrito algunos casos con bipertiroidismo (YETTRA y STARR,50 1951; LAGEZE, CHASSAGNON, MArTR.E-PIERRE 1111 1955), distrofia adiposogenital (BORST y REVERS,10 1949), acromegalia (FALCONER, CoPE y RoBB-SMITH,18 1942) y diabetes insulinorresistente (PEcK y SAGE,'1 1944 ).
En 14 de 35 casos revisados por FERRANTE ¡g (1956) los signos de pubertad precoz aparecieron antes de los dos años de edad.
Cuando se afecta el varón, la precocidad sexual es de observación mucho menos frecuente; sólo existía en cinco de los casos revisados por PRITCHARD ~ ( 19 51). Se han descrito algunos casos con ginecomastia o con distrofia adiposogenital.
EXAMEN RADIOGRÁFICO. - En estos pacientes hay que efectuar el examen radiológico de todo el esqueleto. Se observan las siguientes alteraciones:
En los casos poliostóticos existe clara tendencia a la unilateralidad, siendo las lesiones más acentuadas en los segmentos óseos proximales (fémur y húmero), para ir decreciendo en sentido distal.
Los huesos largos se afectan con mayor frecuencia que los planos y los cortos. El hueso más corrientemente afectado es el fémur, al que siguen, en orden decreciente de frecuencia, según LICHTENSTEIN y JAFFÉ as ( 1942), la tibia, el húmero y el radio.
La zona que en los huesos largos se afecta con mayor frecuencia

152 ANALES DE 1\'lEDICINA
es la metáfisis proximal; luego viene la diáfisis; raras veces la epífisis. Las articulaciones nunca se afectan.
Los huesos planos se alteran por el siguiente orden de frecuencia decreciente: cráneo, pelvis y costillas.
Las lesiones que se observan son en todo superponibles a las de la displasia fibrosa de Jaffé y Lichtenstein:
a) Imágenes geódicas más o menos ovaladas, en cuyo interior ha desaparecido la estructura normal del hueso; que queda sustituida por una imagen homogénea, en general más radiotransparente que el hueso normal.
b) Adelgazamiento e insuflación de la cortical. e) Alternancia de zonas claras con zonas excesivamente conden
sadas . Este aumento de densidad es sobre todo frecuente en la base del cráneo, donde puede llegar a crear problemas de compresión de los pares craneales.
d) Deformidad e incurvaciones de los huesos. Esta deformidad es muy frecuente en la extremidad superior del fémur, que se incurva en <<cayado de pastor» o en «báculo de obispo>>.
e) Cavidades quísticas que, en las manos, recuerdan extraordinariamente la imagen de los encondromas, de los que deben ser diferenciados.
f) Las imágenes geódicas tienen a veces un reborde más denso que las rodea, como si se hubiesen querido retocar repasando con lápiz los contornos de las mismas.
g) El interior de las cavidades tiene a veces una trabeculación fina. En el calcáneo, estas cavidades con trabeculación subtalámica recuerdan las imágenes de la xantomatosis ósea, una de cuyas localizaciones preferentes es precisamente el calcáneo. De hecho, con cierta frecuencia, la biopsia demuestra la presencia en el interior de las cavidades, de células xantomatosas. Se sabe que existen formas de transición entre el síndrome de Albright y las xantomatosis, lo cual, por otra parte, no tiene nada de sorprendente si se recuerda el carácter pluripotencíal de la célula conjuntiva joven.
h) Conservación de extensas zonas de hueso normal, lo cual constituye un dato de gran interés diagnóstico con las osteopatías generalizadas.
Características radiológicas de las lesiones en los diversos tipos de hueso.- Las lesiones ofrecen distintos aspectos radiológicos según asienten en huesos largos, planos o cortos:
1) Huesos largos. En ellos se observan imágenes geódicas redondas u ovales, de contorno trazado a lápiz, cuyo centro es radiográficamente homogéneo, claro, surcado a veces por finas líneas óseas que cuando son muy abundantes pueden conferir a la cavidad un aspecto

P!ULACHS Y COLS. SÍNDROME DE ALBRIGHT 153
en panal de abejas. Al crecer la geoda, la cortical se adelgaza y puede quedar reducida a una fina cáscara que fácilmente se fractura. Presentan también insuflación y abombamiento, incurvaciones y acortamiento del hueso. Es típica la ausencia de reacción perióstica.
2) Huesos ptanos. a) En el macizo facial se producen abombamientos, generalmente unilaterales, de los maxilares, opacificación de los senos paranasales y alteraciones dentarias. Las imágenes radiológicas recuerdan las de las leontiasis ósea y, de hecho, la cara totna un aspecto leonino o acromegaloide; b) En el cráneo hay que distinguir entre la bóveda y la base. En la bóveda se ven imágenes de condensación y engrosamiento de aspecto pagetoide, o, por el contrario, imágenes únicas o múltiples de geodas, de predominio unilateral. En la base, las imágenes son de engrosamiento óseo, con aumento de la densidad, que puede alcanzar aspecto ebúrneo, llegándose a opacificar las células mastoideas. LAYAN! v DURUPT 32 (1951) resumen las anteriores características diciendo que el hemicránea afecto ofrece aspecto pagetoide, con eburnización de la base craneal y leontiasis del macizo facial; e) Del resto de los huesos planos el que más frecuentemente se afecta es el ilíaco, que se llena de imágenes quísticas. Las costillas se afectan raramente, y en el esternón, de modo excepcional. La radiografía del tórax demuestra una escoliosis marcada, que es una expresión más de las deformidades consecutivas a la disminución de la resistencia ósea.
3) Los buesos cortos del carpo y tarso dan imágenes en pequeñas geodas. El calcáneo adopta un aspecto radiográfico «en criba». En las vértebras se observan también imágenes geódicas o en estrías lineales que evocan las osteopatías por carencia.
DIAGNÓSTICO DIFERENCIAL.- El diagnóstico diferencial del síndrome de Albright debe establecerse con las afecciones pertenecientes a dos grandes grupos:
A) AFECCIONES SIN MANCHAS CUTANEAS, pero que pueden present.lt alguna discromia cutánea coexistente:
1) Dis piasia fibrosa de los huesos: se caracteriza por la ausencia de manchas cutáneas y de pubertad precoz.
2) Enfermedad ósea de Recklinghause11: se manifiesta por descalcificación total dd esqueleto, con desaparición, de la lámina compacta; ausencia de zonas hipetostóticas; síndrome bioqtúmico típico de hipercalcemia, hipercalciuria, hiperfosfatemia, hiperfosfatasemia.
3 Enfermedad ósea de Paget: aparece en sujetos de edad avanzada (60-70 años); el aspecto de sus lesiones óseas es característico: engrosamiento de la cortical, muy típico, y estructura filamentosa o algo-

154 ANALES DE .M~EDICINA
donosa; no existen manchan cutáneas; hay gran elevación de las fosfatasas alcalinas.
4) Mieloma: faltan las manchas cutáneas; imagen radiológica de geodas en sacabocados, sin ribete condensante; hiperproteinemia, hiperproteinuria de Bence-Jones; abundantes plasmocitos en la punción esternal.
5) Discondroplasia de Ollier: afecta únicamente a las metáfisis de los huesos largos y a los huesos cortos de las manos y de los pies.
B) AFECCIONES CON MANCHAS CUTANEAS 1) N eurofibromatosis: las manchas de la piel ofrecen los caracte
res diferenciales que ya antes hemos señalado; hay coexistencia de tumotes nerviosos y de tumores cutáneos.
TRATAMIENTO.- Se ha aconsejado la radioterapia; quizá pueda tener cierta eficacia cuando las lesiones son localizadas.
Se han utilizado el fósforo y la vitamina D, con nula eficacia terapéutica. HELPET 2
; (1940) señala haber obtenido algún resultado favorable con acetato o gluconato de alúmina, administrados con objeto de precipitar el fósforo intestinal. Este método ha sido usado por otros autores sin conseguir éxito terapéutico alguno.
CALDWELL y BRODERICK 12 ( 1948) trataron a una paciente, una niña
negra de 9 años, con acetato de alúmina por vía bucal, siguiendo el concepto de HELPET 27 (1940) que consideraba que dicha solución, al precipitarse en forma de fosfatos en la luz intestinal, podría reducir la absorción de fósforo, con el subsiguiente aumento de retención de calcio. Se preparan las siguientes soluciones:
Solución de subacetato de alúmina Acido acético glacial Agua, s. c. para .
545 c.c. 15 c.c.
1.500 c.c.
No consiguieron objetivar resultados favorables en las radiografías, ni obtener el menor cambio en los valores químicos de la sangre.
No obstante, GHORMELEY e HINCHLEY 22 ( 1944) han conseguido me
jorías radiológicas evidentes, de aumento de la calcificación, en varios casos de enfermedad malácica de los huesos.
Dada la falta de fundamen to de estos tratamientos, así como la ineficacia de los mismos, creemos que la terapé;.¡tica del síndrome de Albright debe actualmente limitarse al tratamiento sintomático de las complicaciones óseas que pueda presentar, es decir, las deformaciones y, sobre todo, las fracturas .

PIUI.ACHS Y COLS. SÍNOROi\IE DE ALBRIGH T 155
RESUMEN. - Los autores describen un caso de síndrome de Albright en un paciente de 24 años de edad y a propósito del mismo señalan los caracteres clínicos y radiográficos que permiten el diagnóstico.
Cátedra de Patología Quirúrgica II de la Facultad de Medicina de Barcelona.
Prof. P. PruLACHS.
BIBLIOGRAFIA
ALBRIGil'f, F.: Polyostot ic fibrous dysplasia: a defense of tbe entity. J. Clin. Endocrino!., 7, 307, 1947. ALBR!GIIT, F., BuTLER, A. M., IiM.JPTON, A. D. y s~nTil, P.: Syndrome characrerized by osteítis fibrosa disseminatn; areas of pigmentation and endocrine dysfunction; with prccocious puberty in fernales: Report of Jive cases. New England J. M., 216, 727, 1937. AJU.I:EN·SOBORG, U. y IvERSEN, T.: Albrigh1's syndrome. Acta pediátrica, 45, 558, 1956. BASl'EDO, D. L. A.: Coexistence of Albright's syndrome and mongolism. Canad. Med. Ass. J., 84, 1135, 1961. BEilREND, A.: Síndrome de Albrigbt. Presentación de un caso con fracturas patológicas múltiples, fibrosis ósea diseminada, pubertad precoz y nevos pigmentados múltiples. An. Cir. (ed. esp.), 4, 247, 1945. BEI\lilliCT, P. H.: Sex precocity and polyostotic fibrous displasia; repon of n case in a hoy wíth testicular biopsy. Amer. J. Dis. Child., 11, 426, 1966. B.ENEDIC'f, P. H ., SZAnó, G., FrTZPA'I'RJCK, T. B. y $1NESI, S. J.: Melanotic macules in Albright's syndrome and in neuroJibromatosis. J.A.M.A., 205, 618, 1968. BERARDINELLI, W.: Two cases of Albright's syndrome observed in Brazil. J. Clin. En-docrino!., 10, 1499, 1950. Be:RNÁI.DEZ SARMIENTO, P.: Síndrome de Albright. Rev. Ortop. y Traumatol., 2, 27, 1958. BoRTs, W. H. y REVERS, F. E.: Albright's disease. Acta med. Scandinava, 135, 91, 1949. BUKER, R. H., I!UGRES, F. A. y MASHliURN, J. D.: Polycstotic fibrous displasia (Albright's syndrome). J. Thor. Cardiov. Surg, 49, 241, 1965. CALDWELL, G. A. y BRODERICK, T. F.: Polyostotic fibrous dysplasia in one of negro twin girls. Ano. Intem. Med., 27, 114, 19.J7. CAPLAN, D. B. y FEINGOLD, M.: Polyostotic fibrous dysplasia with cutaneotrs pigmentatino. Amcr. J. Dis. Child., 109, 575, 1965. CARLETTI, B. y KEHYAYAN, E.: Un caso di sindromi di Albright. Minerva Pediatrica, 14, 159, 1962. Docr-.'ERTY, M. B., GnoRMLEY, R. K., KENNEDY, R. L. J. y PuGil, D. G.: Albright's syndrome (polyostotic fibrous dysplasia with cutaneous pigmentation in both sexes and gonadal dysfunction in females). Arch. Int. Med., 75, 357, 1945. DoCKERTY, M. B., MEYERDING, H. W. y WALLACe, G. T.: Albright's Syndrome: (fibrous dysplasia of bones with cutaneous pigmentation in botb sexcs and gonada l dysfunction in females). Proc. Staff. Mcet. Mayo Clinic, 19, 81, 1944. DuCROQUET, R. J. P.: Traitement chirurgical des localizations f~morales de 6 cas de syn· drome d'Albright. Press. méd., 60, 602, 1952. FALCONER, M. A., CoJ>E, C. L. y RoBB·S~uTH, A. H. T.: Fibrous dysplasia ol bone with endocrine disorders and cutaneous pigmentation (Albright's disease). Quarr. J . Med., 11, 121, 1942. FERRAN1'E, L.: 11 quadro precoce della síndrome di Albri¡¡ht. Acta pediat. Jat., 9, 129, 1956. FON'fAINE, R., WARTER, P., MULLlR, J. N., $TOLL, G. y GANDAR, P.: Ost~ite Jibro-géodique disseminée a predominance unilatérale avec pigmentation cutanéc et puberté pr6-coce. ]. Radio!. Elcctr., 35, 893, 1954. GALI.ICO, E. y MARKOVJTS, S.: Contributo allo studio dalla síndrome di Albright. ?vliner· va med., 57, 118, 1966. Gno~rLEY, R. K. y HINCHEY, J. T.: Use of aluminum acetate in tbe treaternent of malacic diseases of bone. J. Bone & Joint Surg., 26, 811, 1944.

156 ANALES DE MEDICINA
GlRAUO, ].: Contl'ibution a l'étude du syndrome d'Aibright. These, Lyon, 1959. GoRIIAM, L. W., CAMPBELL, E . H., HowARD, W. P., DoNRUS ER, }. L . y Rusr, N. H .:
Albright's syodrome: A group of cases characterized by osteitis fibrosa clisscminata, areas of pigmentation and a ganada! dysfunction. Clinics , 1, 358, 1942.
HACKETT, L. J., jc. y CHRISTOPHERSON, W. M.: Polyostoric fibrous dysplasia. ]. Pediat, 35, 767, 1949.
HALL, P.: Albright's syndrome in an adult male. Report of an atypicnl case ith psychiatric symptoms. Brit. Med. J., 2, 1159, 1962.
I!ELPET, A. J.: A new conceprion of parnthyroid function and its dinical application: A preliminary repon on resulrs of treatrnent of generalized fibrocysric and allied bone diseases and of rheumatoid arrhritis by aluminum acetare. Brit. }. Surg., 27, 651, 1940.
HERNBERC, C. A. y EDGREN, W.: Morbus Albright-Jaffé-Lichtenstein, osreolibrosis deformans juvenilis. Acra med. Scandinav., 135, 208, 1949.
LACEZE, P., CRASSAGNON y MAtTREPIERRE, J. : Syndrome d'Albright et hiperthyroid (a propos d'une observation). Lyon méd., 87, 200, 1955.
LAPORTE, F . y LÉCER, H.: Le syndrome d'Albrigbt . J Chir., 82, 457, 1961. l.ARA, L. DE, CASTTLLO, G. y FRANCO, R.: Síndrome de Albright. Rev. clin. esp., 34, 1,
1949. LAYANI, F. y DuRUPT, L.: Dysplasic fibrcusc des os (maladie e }affé-Lichtenstein, syndro
me d'Aibright). Encycl. Méd. Chir. Os-Articulations T: 1, 14024, F-10, 1-10, París, 1951.
LÉGER, L., DuCROQUET, R. y LÉGER, H.: Maladies du squelette. Masson Ed., París, 1940. LtCHTENSTETN, L.: Polyoctotic úbrous dysplasia. Arch. Surg., 36, 874, 1938. LtCIITENSTEIN, L. y JAFFÉ, H. L.: Fibrous dysplasia of bone. Arch. Path., 33, 777, 1942. MAcCuNE, D.: Osteítis fibrosa cystica; the case of a nine years old firl who also exhibits
precocious puberty, multiple pigmentation of the skin and biperthyroidism. Transacrions of tbe Sociery for Pediatric Research, Annual Meeting, May, 5, 1936. Am. J . Dls. Cbild., 52, 743, 1936.
MACCUNE, D. y BRUCR, H .: Osteodystrophia fibrosa. Report of a case in whicb the condition was combined with precocious puberty, pathological pigmentation of the skin and hyperthyroidism with a revue of rhe literature. Am. J. Dis. Child., 5-I, 806, 1937.
MAGGI, R. y RAVERA, }. }.: Displasia 6sea, pubertad precoz y manchas melanodérmicas (Síndrome de Albright). Arch. Ped. Urug., 41, 29, 1970.
MARTiN, A.: La displasia fibrosa poliost6tica o enfermedad


















