
SR1 en los siguientes casos. Molécula 1024(cm3) … · Ar 1.63 363.3 87.3 Kr 2.46 322.7 119.9 Xe...
27
SR1.- Calcular la energía de interacción de dispersión para dos moléculas situadas a 5 Å, en los siguientes casos. Molécula ’10 24 (cm 3 ) I(Kcal/mol) T eb (K) Ne 0.392 497.2 27.3 Ar 1.63 363.3 87.3 Kr 2.46 322.7 119.9 Xe 4.01 279 165.1 Comparar el resultado obtenido con la temperatura de ebullición normal. Para un par de moléculas no polares el único térmico atractivo es el de dispersión 6 2 1 2 1 2 1 di di r ' ' I I I I 2 3 V Si la molécula 1 y 2 son iguales 6 2 di di r ' I 4 3 V a) Ne-Ne 6 10 3 3 30 18 di di m 10 5 m 10 392 . 0 J 10 454 . 3 4 3 V 1 23 mol · kJ 015 . 0 J 10 548 . 2
Transcript of SR1 en los siguientes casos. Molécula 1024(cm3) … · Ar 1.63 363.3 87.3 Kr 2.46 322.7 119.9 Xe...

SR1.- Calcular la energía de interacción de dispersión para dos moléculas situadas a 5 Å,
en los siguientes casos.
Molécula ’1024(cm3) I(Kcal/mol) Teb (K)
Ne 0.392 497.2 27.3
Ar 1.63 363.3 87.3
Kr 2.46 322.7 119.9
Xe 4.01 279 165.1
Comparar el resultado obtenido con la temperatura de ebullición normal.
Para un par de moléculas no polares el único térmico atractivo es el de dispersión
6
21
21
21didi
r
''
II
II
2
3V
Si la molécula 1 y 2 son iguales6
2
didir
'I
4
3V
a) Ne-Ne
610
333018
didi
m105
m10392.0J10454.3
4
3V
123 mol·kJ015.0J10548.2

Moléculas Vdi-di(kJ·mol-1) Teb(K)
Ne-Ne -0.015 27.3
Ar-Ar -0.19 87.3
Kr-Kr -0.39 119.9
Xe-Xe -0.90 165.1
A mayor interacción atractiva entre las moléculas mayor es la energía cinética
(temperatura) necesaria para vencer las interacciones intermoelculares y pasar del
estado líquido al gaseoso

SR2.- Justificar, para el caso del Argón en el problema anterior, por qué se ignoran habitualmente las fuerzas gravitacionales. Considerar que la
distancia mínima de acercamiento, dada por el diámetro molecular, es de 5.72 Å.
La energía de interacción tiene la forma6didi
r
CV
Por lo que la fuerza entre las moléculas debida a la interacción atractiva será
76didididir
C6
r
C
rVF
Siendo C2'I
4
3C Con los datos del problema anterior, para el caso del Argón 678 m·J10·03.5C
A una distancia de r=5.72 Å=5.72·10-10 m la fuerza será N10·506.1m·J10·506.1F 1212
didi
El valor negativo indica una fuerza atractiva, que intenta disminuir la distancia entre las moléculas.
La fuerza de interacción gravitatoria entre dos moléculas de Argón es:
221
gr
mmGF Siendo m1=m2=(39.95·10-3/NA ) Kg y G=6.67·10-11 Nm2Kg-2
Resultando que a la distancia de 5.72·10-10 m la fuerza gravitatoria es N10·97.8F 43
g
Es decir, la fuerza de origen electrostático entre las moléculas es del orden de 1030 veces más
grande que la fuerza gravitatoria. O, lo que es lo mismo, a todos los efectos podemos
despreciar esta última.

SR3.- Sabiendo la polarizabilidad del Argón (problema 1) y el momento dipolar del agua (1.85 Debyes), calcular a) la
energía de interacción dipolo-dipolo inducido a la distancia de contacto. Para ello considerar ambas moléculas como
esferas con diámetros rígidos de 3.08 Å para el argón y 2.76 Å para el agua. b) Si la polarizabilidad de volumen del agua
es de 1.45 10-30 m3 y los potenciales de ionización del agua y argón son respectivamente 12.6 y 15.8 eV ¿cuál es la
energía de interacción atractiva total ?.
Ar H2O
1 2
r = 1/2 + 2/2 = 2.92 Å
a) La energía de interacción dipolo-dipolo inducido es:
6
0
1
2
26
0
2
2
1did
r4
'
r4
'V
Como el dipolo de la molécula 1 (Ar) es cero queda simplemente:
122
61011212
330230
6
0
1
2
2did
mol·kJ54.0J10·00.9
m10·92.2mJC10·8542.8·4
m10·63.1m·C10·17.6
r4
'V

b) Para tener la energía de interacción atractiva total hemos de sumar la interacción dipolo
inducido-dipolo inducido (la dipolo-dipolo es nula porque el momento dipolar del Ar es cero)
6
21
21
21didi
r
''
II
II
2
3V
121
610
3030
1818
1818
didi mol·kJ87.3J10·43.610·92.2
10·63.1·10·45.1
10·531.210·019.2
10·531.2·10·019.2
2
3V
Utilizando el sistema internacional (pasando la energía de ionización a Joules, las
polarizabilizades a m3 y la distancia a m) queda:
1
diddidiatrac mol·kJ41.454.087.3VVV

SR4.- Suponga que la interacción entre dos moléculas tiene la forma V(r)=B/rm –C/rn. Mostrar
que una condición necesaria para que V(r) tenga un mínimo es que las fuerzas repulsivas sean
de menor alcance que las atractivas (es decir, que m>n).
nm r
C
r
B)r(V
La condición de mínimo es
0dr
)r(dV
0rr
0dr
)r(Vd
0rr
2
2
Si hacemos la derivada primera e igualamos a cero:
1n1m r
B·n
r
C·m
dr
)r(dV
0dr
)r(dV 1n
0
1m
0 r
B·n
r
C·m
mn
1
0C·m
B·nr
Si hacemos la derivada segunda:
1n1m2
2
r
B·n
r
C·m
dr
d
dr
)r(Vd
2m2n2
2
r
)m1(C·m
r
)n1(B·n
dr
)r(Vd
Nótese que para que haya un punto singular (mínimo, máximo o de inflexión) n≠m

Si calculamos la derivada segunda en el punto singular r0
mn
2m
mn
2n
rr
2
2
C·m
B·n
)m1(C·m
C·m
B·n
)n1(B·n
dr
)r(Vd
0
Operando se llega a la expresión:
mnC·mB·ndr
)r(Vdmn
2m
mn
2m
rr
2
2
0
Si n>m 0dr
)r(Vd
0rr
2
2
y r0 es un mínimo
Si n<m 0dr
)r(Vd
0rr
2
2
y r0 es un máximo

SR5.- El potencial de Lennard-Jones tiene la forma general VLJ=-C/r6 + B/r12 . Expresar el potencial en términos de (valor de la
energía en el mínimo de la curva de interacción) y r0 (distancia de separación en el mínimo), así como de y (distancia cuando
VLJ=0).
-1
2,5
VLJ
r
1/r12
-1/r6
r0
-0
612LJr
C
r
B)r(V
r;r,VV 0LJLJ
Supongamos que queremos expresar VLJ en función de la posición del mínimo y la profundidad
del pozo
r0 es la posición del mínimo 0dr
)r(dV
0rr
LJ
es el valor de r que hace VLJ=0
= -VLJ(r=r0)
0dr
)r(dV
0rr
LJ
El mínimo lo podemos encontrar tomando la primera derivada de (1) e igualando a cero
(1)
13
0
7
0 r
B12
r
C6
137
LJ
r
B·12
r
C·6
dr
)r(dV
6/1
0C
B2r
(2)

La profundidad del pozo en el mínimo la podemos encontrar sustituyendo (2) en (1) y
cambiando el signo
)rr(V 0LJ
B4
C
C
B2
C
C
B4
B
r
C
r
B 2
2
26
0
12
0
Tenemos por tanto dos ecuaciones ((2) y (3)) que relacionan B y C con r0 y
(3)
B4
C2
6/1
0C
B2r
6
0
12
0
r2C
rB
Y sustituyendo en (1)
6
6
0
12
12
0
612LJr
r2
r
r
r
C
r
B)r(V
6
0
12
0LJ
r
r2
r
r)r(V

r;,VV LJLJ
Supongamos que queremos expresar VLJ en función de la posición que anula elpotencialy la
profundidad del pozo
El valor de r que anula VLJ lo podemos obtener fácilmente
612
CB0
6/1
C
B
Tenemos por tanto dos ecuaciones ((2) y (4)) que relacionan B y C con y
(4)
B4
C2
6
12
4C
4B
6/1
C
B
Y sustituyendo en (1)
6
6
12
12
612LJr
4
r
4
r
C
r
B)r(V
612
LJrr
4)r(V

SR6.- Calcular la fuerza entre dos moléculas de metano separadas a) 8 Å b) 5 Å y c) 3 Å si la interacción obedece a un potencial de Lennard-
Jones con parámetros = 0.013 eV y =3.8 Å. d) ¿Cuándo se anula la fuerza?
612LJr
C
r
B)r(V
intVF
7
6
13
12
LJ
rr
224
dr
)r(dVF
a) N10·01.7Å·eV10·38.4
8
8.3
8
8.32013.0·24Å)8r(F 131-4
7
6
13
12
b) N10·18.1Å)5r(F 11
c) N10·00.5Å)3r(F 9
d) 0rr
2240F
7
6
13
12
7
6
13
12
rr
2
Å265.4·2r 6/1
V
ra
b
c
d

SR7.- La energía de interacción del sistema H2O-CCl4 en el mínimo de la curva de Lennard-Jones es de –0.10 kJmol-1. Determinar la
distancia de equilibrio teniendo en cuenta que las interacciones atractivas de este sistema son la suma de interacciones dipolo-dipolo inducido
y dipolo inducido-dipolo inducido. Datos:
Molécula ’1024(cm3) μ (D) I (eV)
H2O 1.45 1.85 12.6
CCl4 11.2 0. 11.5
612atracrepLJr
C
r
B)r(V)r(V)r(V
6
0
12
0LJ
r
r2
r
r)r(V
6/1
0
6
02
Crr2C
Conocido podemos obtener r0 si calculamos primero C:
dididid6atrac VVr
C)r(V
Como la molécula de CCl4 es apolar no hay interacción dipolo-dipolo y la interacción d-di será
entre el dipolo del agua (1) y el inducido en el tetracloruro de carbono (2)
621
21
216
0
2
2
1dididid6 r
''
II
II
2
3
r4
'VV
r
C
Con lo que C queda:
''II
II
2
3
4
'C 21
21
21
0
2
2
1

Sustituyendo datos:
m·C10·17.6 30
1
330
2 m10·2.11'
330
1 m10·45.1'
J10·02.2I 18
1
J10·84.1I 18
2
1112
0 mCJ10·85419.8
677 m·J10·73.2C
Å59.6m10·59.62
Cr 10
6/1
0

SR8.- Conocidos los valores para la densidad de la trimetilamina gaseosa en función de la presión, a la temperatura de 0 ºC:
P(atm) 0.2 0.4 0.6 0.8
ρ(gl-1) 0.5336 1.0790 1.6363 2.2054
Calcular el segundo coeficiente del virial y el peso molecular.
Supondremos que dado que trabajamos a presiones moderadas, la ecuación de estado del
gas podrá aproximarse como:
m
m
V
)T(B1
RT
PV
Dado que los datos vienen en función de la densidad podemos efectuar el cambio
(1)
M
·n
m
n
VVm
Con lo que la ec. (1) queda como:
M
)T(B1
RT
PM
Y reordenando:
)T(BM
RT
M
RTP2
(2)
Es decir, una representación de P/ frente a debería dar una línea recta de cuya ordenada en
el origen podemos obtener M y de la pendiente B(T)

0.3748 0.5336
0.3707 1.079
0.3667 1.6363
0.3627 2.2054
P/ (atm·l·g-1) (g·l-1)
y = -0.0072x + 0.3786R² = 0.9998
0.362
0.364
0.366
0.368
0.37
0.372
0.374
0.376
0 0.5 1 1.5 2 2.5
(g·l-1)
P/
(atm
·l·g
-1)
Comparando el resultado del ajuste con (2) tenemos:
1g·l·atm3786.0M
RT
22
2g·l·atm0072.0)T(B
M
RT
Sabiendo que R=0.082 atm·l·K-1·mol-1 y T=273.15 K,
obtenemos:
1mol·g16.59M
1mol·l1297.1)T(B
Datos experimentales:

SR9. Para estudiar el comportamiento de una determinada sustancia a T=65 K se necesita
describir las interacciones que aparecen entre las moléculas que la forman. Para ello se elige
un potencial de la forma
r
r
Cr
)r(V6
V
r
a) Determinar el valor de C de la anterior expresión teniendo en cuenta las siguientes
propiedades de las moléculas.
(D) ’(cm3) I (kcal/mol)
0.4 1.5 10-24 50
66
21
21
21
6
0
1
2
2
6
0
2
2
1
62
0
2
2
2
1didididddatra
r
C
r
''
II
II
2
3
r4
'
r4
'
r4KT3
2VVVV
''
II
II
2
3
4
'
4
'
4KT3
2C 21
21
21
0
1
2
2
0
2
2
1
2
0
2
2
2
1
2
0
2
2
0
4
'I4
3
4
'2
4KT3
2C
C=8.24410-79 Jm6

b) Para un amplio intervalo de presiones, el comportamiento en fase gas de esta sustancia
viene adecuadamente descrito por la siguiente función de estado:
m
m
V
)T(B1
RT
PV
Sabiendo que el gas muestra un comportamiento ideal a T=65K (temperatura de Boyle)
determinar el valor de en la expresión del potencial intermolecular.
(Nota: suponer que a distancias mayores que kT >> V(r))
A T=65 K B(T)=0 drre1N2)T(B 2
0
kT)r(V
A
int
V
r
dre1rN2)T(B0
kT)r(V
2
A
int
dre1rdre1rN2 kT)r(V
2
0
kT)r(V
2
A
intint
dre1rdre1rN2 kTrC
2
0
kT2
A
6
drkTr
C11rdr01rN2
6
2
0
2
A
3
A
0
3
Ar3
1
kT
CN2
3
rN2
3
A
3
A
kT3
CN2
3
N2
A T=65 K B(T)=0
3
A
3
A
kT3
CN2
3
N2)T(B
3
A
3
A
kT3
CN2
3
N2
=3.12 Å

V(R)
Rσ1
σ2
0
∞
-
SR10.- Calcular la expresión del segundo coeficiente del virial para un gas cuyo potencial de interacción
tiene la forma:
El potencial de interacción intermolecular tiene la forma:
12
221
2
1
)r()r(Vr
0Vr
Vr
)r(V
Ecuación de la recta que pasa por los puntos
(2,0) y (1,-)
12
1
12
1
yy
yy
xx
xx
Siendo (x1,y1) = (2,0) y (x2,y2) = (1,-)
Con este potencial podemos calcular ahora el coeficiente del virial B(T) haciendo uso de
la ecuacióndrre1N2)T(B 2
0
kT)r(V
A
int

dre1rN2)T(B0
kT)r(V
2
A
int
dre1rdre1rdre1rN22
int2
1
int1 intkT
)r(V2kT
)r(V2
0
kT)r(V
2
A
dre1rdre1rdre1rN22
2
1
12
2
1KT
02)(kT
)r(
2
0
kT2
A
0dre1rdrrN22
1
212
1)r(
)(kT2
0
2
A
dre1rN23
N2 2
1
212
)r()(kT2
A
3
1A
La integral que queda puede resolverse fácilmente considerando que kT >> V(r). En ese
caso:)r(
)(kT1e 2
12
)r()(kT
212
dr)r(
)(kT11rN2
3
N2)T(B
2
12
12
2
A
3
1A
drr
)(kT
N2drr
)(kT
N2
3
N2 2
1
2
1
2
12
2A3
12
A
3
1A
3)(kT
N2
4)(kT
N2
3
N2 3
1
3
2
12
2A
4
1
4
2
12
A
3
1A
El coeficiente B(T) queda en este caso:

SR11.- Considere un cristal de N átomos. La energía vibracional de cada modo normal puede ser escrita como =xh, donde x
puede tomar cualquier valor continuo entre 0 y +. Además, como en el tratamiento de Einstein sólo es posible una única
frecuencia vibracional . Calcular: a) la función de partición del cristal, b) la energía vibracional c) la capacidad calorífica del
cristal.
Para un sólido de N átomos 3·Ngrados de libertad.
3 rotaciones y 3 traslacion,es así que habrán 3·N-6vibraciones
Si N es muy grande 3·N-6 será aproximadamente igual a 3·N
Tomando las 3N vibraciones como independientes (modos normales) la función de partición del
sólido será:
N3
1i
i,vibvib qQ
Nos dice el enunciado que cada una de las vibraciones tiene una energía que puede variar de
forma continua, por lo que la función de partición de cada una de ellas queda:
0
xh
j
i,vib dxeeq j
Es decir, la suma sobre un conjunto discreto de estados se transforma en una integral ya que
el ‘número cuántico’ x varía de forma continua. La integral se resuelve de forma inmediata:
h
kT
h
1e
h
1q 0
xh
i,vib
Como todas las vibraciones tienen la misma frecuencia la función de partición canónica del
sistema queda:
N3N3
1i
N3
1i
i,vibvibh
kT
h
kTqQ

La contribución vibracional a la energía interna será:
NkT3dT
h
kTlnd
NkT3T
h
kTln
kTT
QlnkTU 2
V,N
N3
2
V,N
vib2
vib
La contribución vibracional a la capacidad calorífica será:
Nk3
T
NkT3
T
UC
V,NV,N
vibvib,V
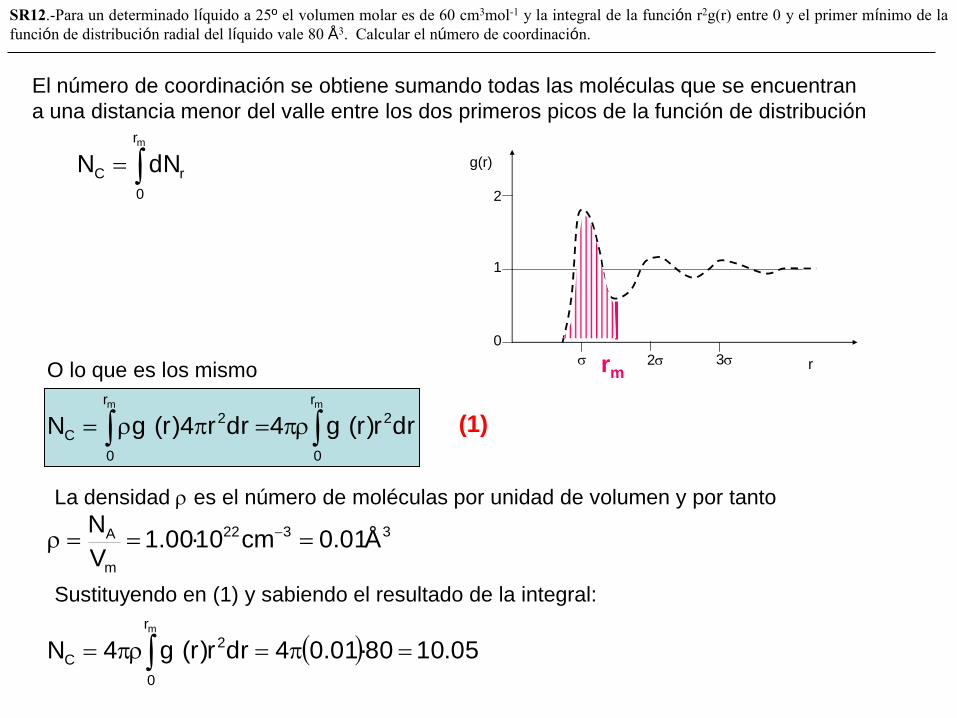
SR12.-Para un determinado líquido a 25º el volumen molar es de 60 cm3mol-1 y la integral de la función r2g(r) entre 0 y el primer mínimo de la
función de distribución radial del líquido vale 80 Å3. Calcular el número de coordinación.
mr
0
rC dNN
2 3
g(r)
r
1
0
2
mm r
0
2
r
0
2
C drr)r(g4drr4)r(gN
rm
El número de coordinación se obtiene sumando todas las moléculas que se encuentran
a una distancia menor del valle entre los dos primeros picos de la función de distribución
O lo que es los mismo
La densidad es el número de moléculas por unidad de volumen y por tanto
3322
m
A Å01.0cm10·00.1V
N
05.1080·01.04drr)r(g4Nmr
0
2
C
Sustituyendo en (1) y sabiendo el resultado de la integral:
(1)

SR13.- La siguiente figura muestra las funciones de distribución radial, calculadas mediante dinámica molecular para el anión cloruro
en agua (Cl-O y Cl-H). A partir de ellas deducir cómo se disponen, en término medio, las moléculas de agua en la primera capa de
solvatación. ¿Cómo podría calcularse a partir de la función de distribución el número de moléculas que forman esta primera capa de
solvatación?
2.4 Å
3.3 Å
3.6 Å
La función de distribución radial de los hidrógenos presenta dos picos, situados a 2.4 y
3.6 Å, mientras que la de los oxígenos presenta un solo pico situado a 3.3 Å, es decir 0.9
Å más alejado que el primer pico de los hidrógenos. Este hecho quiere decir que las
moléculas de agua solvatan al ión cloruro encarando los hidrógenos hacia el ión, como
corresponde a una interacción entre la carga negativa y el dipolo del agua.

Además, si tenemos en cuenta que la molécula de agua presenta la siguiente geometría:
~0.95 Å
~105º
Podemos comprobar que la diferencia entre las posiciones más probables de los
hidrógenos y oxígenos de la primera capa coinciden con la longitud del enlace O-H. Es
decir, tendremos necesariamente una disposición lineal entre los átomos Cl(-)·····H-O
Cl- H O
~ 180º
H
Para confirmar el modelo podemos calcular la distancia del cloruro al segundo hidrógeno
y compararla con el segundo pico de la función de distribución
Cl- H O
H~ 105ºa?
b = 3.3 Å
c = 0.97 Å

El lado ‘a’ se obtiene por el teorema del coseno:
cosbc2cba 222
Tomando c=0.97 Å, b=3.3 Å y =105º se obtiene a=3.6 Å, en acuerdo con la
observación de la función de distribución radial.
mm r
0
2
r
0
2
C drr)r(g4drr4)r(gN
El número de coordinación se obtiene sumando todas las moléculas que se encuentran
a una distancia menor del valle entre los dos primeros picos de la función de distribución
En este caso podemos hacerlo con la función de distribución radial de los oxígenos o de
los hidrógenos.
Nota: las funciones de distribución radial presentadas en la figura se han normalizado
respecto a la densidad de moléculas de agua. Como hay 2 hidrógenos por molécula, la
función de distribución de los hidrógenos tiende a 2 a largas distancias en lugar de a
uno.

SR14.- En la siguiente figura se dan las funciones de distribución para el catión Na+ en agua (Na-O y Na-H). Deducir la disposición de las
moléculas de agua en la primera capa de solvatación. Entre el primer y el segundo pico de la función Na-O aparece un mínimo muy
pronunciado, ¿cuál sería el significado físico de que este mínimo llegase a g(r)=0?, ¿en qué casos puede esperarse este comportamiento?.
2.5 Å3.1 Å
5.4 Å
4.9 Å
En este caso los oxígenos están más cercanos al ión que los hidrógenos, debido a la
carga positiva del mismo. La diferencia entre las posiciones más probables de los
oxígenos e hidrógenos de la primera capa es menor que la distancia de enlace O-H, lo
que indica que NO hay una disposición lineal Na(+)·····O-H
Además el segundo pico de la función de distribución de lso hidrógenos está muy lejos
de la posicíon del primer pico de los oxígenos, lo que indica que estos hidrógenos no
están unidos a los oxígenos d ela primera capa. Corresponden a moléculas de la
segunda capa.

Na+ O
H
2.5 Å
3.1 Å
0.97 Å
Así pues, los dos hidrógenos de las moléculas de agua de la primera capa aparecen a la
misma distancia (en un solo pico) respecto del Na(+)
Na+
O
HH
2.5 Å
3.1 Å
3.1 Å
0.97 Å0.97 Å
El ángulo Na···O-H se puede calcular usando el teorema del coseno:
cosbc2cba 222
Tomando c=0.97 Å, b=2.5 Å y a=3.1 Å se obtiene =125º.
Si la función de distribución se anula entre el primer y segundo pico indica que la
probabilidad de encontrar moléculas de agua es nula a esa distancia. Este fenómeno
aparece en iones que por su gran carga (ej. Al(3+)) o pequeño tamaño (el Be(2+)) atraen
fuertemente a las moléculas de agua de la primera capa no permitiendo que se alejen y
que se intercambien con moléculas procedentes del resto del disolvente. En este caso el
ión se mueve con una esfera de solvatación permanente.


















