
TRABAJO FIN DE ESTUDIOS - Biblioteca de la Universidad de ... · los órganos verdes de la vid,...
103
TRABAJO FIN DE ESTUDIOS Estudio fotolítico del fungicida Ametoctradín Laura Gárriz Soba MÁSTER EN QUÍMICA AVANZADA Tutores: María Teresa Martínez Soria y Jesús Sanz Asensio Facultad de Ciencias, Estudios Agroalimentarios e Informática Curso 2011-2012
Transcript of TRABAJO FIN DE ESTUDIOS - Biblioteca de la Universidad de ... · los órganos verdes de la vid,...

TRABAJO FIN DE ESTUDIOS
Estudio fotolítico del fungicida Ametoctradín
Laura Gárriz Soba
MÁSTER EN QUÍMICA AVANZADA
Tutores: María Teresa Martínez Soria y Jesús Sanz AsensioFacultad de Ciencias, Estudios Agroalimentarios e Informática
Curso 2011-2012

© El autor© Universidad de La Rioja, Servicio de Publicaciones, 2012
publicaciones.unirioja.esE-mail: [email protected]
Estudio fotolítico del fungicida Ametoctradín, trabajo fin de estudiosde Laura Gárriz Soba, dirigido por María Teresa Martínez Soria y Jesús Sanz Asensio
(publicado por la Universidad de La Rioja), se difunde bajo una LicenciaCreative Commons Reconocimiento-NoComercial-SinObraDerivada 3.0 Unported.Permisos que vayan más allá de lo cubierto por esta licencia pueden solicitarse a los
titulares del copyright.

Laura Gárriz SobaJunio 2012
Estudio fotolítico del fungicida Ametoctradín
DEPARTAMENTO DE QUÍMICAUNIVERSIDAD DE LA RIOJAÁREA DE QUÍMICA ANALÍTICA

Doña María Teresa Martínez Soria y Don Jesús Sanz Asensio, Profesores
Titulares de Química Analítica del Departamento de Química de la
Universidad de La Rioja.
Certifican:
Que el trabajo de investigación “Estudio fotolítico del fungicida
Ametoctradín” ha sido realizado por la licenciada Laura Gárriz Soba en los
laboratorios del Departamento de Química de la Universidad de La Rioja
bajo su inmediata dirección y reúne las condiciones exigidas para
conseguir el título de Máster en Química Avanzada.
Logroño, 18 de Junio de 2012
Fdo: María Teresa Martínez Soria Fdo: Jesús Sanz Asensio

Me gustaría desde estas líneas expresar mi agradecimiento a todas las
personas que me han ayudado y colaborado en la realización de este
proyecto.
A mis directores de investigación, Mayte y Jesús por ofrecerme la
oportunidad de realizar este trabajo. Agradecerles también el
conocimiento transmitido durante este tiempo además de su ayuda.
A mis compañeros de laboratorio: Franz, Alejandro, Ana J., Pilar, Cristina,
Fernando, José Miguel, Ana G. y Marivel por compartir conmigo su tiempo
y buenos momentos así como su experiencia y conocimientos, sin los que
no hubiese sido posible adentrarme en esta aventura. También
agradecerles su ánimo y apoyo en todo momento, especialmente cuando
más lo he necesitado.
A Txino, Amaia y el resto de servicio de laboratorios por la ayuda ofrecida.
A Nines y Ernesto, por el tiempo y trabajo dedicado a inyectar mis muestras
así como a resolver los problemas que han ido surgiendo en el UPLC.
Agradecerles también el conocimiento transmitido acerca del equipo.
A Miguel Ángel por resolver mis dudas fotoquímicas.
A mis compañeros inorgánicos y orgánicos del Máster: Sábel, Rocío, Jesús,
Justo, Laura, Claudio e Ismael.
A mis padres y a mi hermana por su cariño y apoyo incondicional; por su
constante estímulo y haber confiado en mí siempre. Sin ellos no hubiese
sido posible llegar hasta aquí.
A mis amigos y a mi novio, por estar siempre a mi lado y su constante
ayuda, paciencia, ánimo y comprensión.
A todos, mil gracias.

ÍNDICE
CAPITULO I: FUNGICIDAS EN VID 5
1.1. Enfermedades de la vid causadas por hongos 7
1.1.1. Oidio 7
1.1.2. Mildiu 8
1.1.3. Botritis o Podredumbre gris 9
1.1.4. Excoriosis 10
1.1.5. Eutipiosis o eutipia 10
1.1.6. Yesca 11
1.2. Productos fitosanitarios 11
1.3. Sistemas de producción integrada 13
1.4. Materia activa estudiada: Ametoctradín 14
1.4.1. Características físico-químicas 14
1.4.2. Perfil toxicológico y ecotoxicológico 16
1.4.3. Modo de acción 17
1.4.4. Caracterización biológica 18
CAPÍTULO II: DEGRADACIÓN DE FUNGICIDAS 21
2.1. Mecanismos de degradación 23
2.2. Cinética de los procesos de degradación 24
2.3. Estudios de fotodegradación 25
2.4. Análisis de residuos de fungicidas en muestras enológicas 29
CAPÍTULO III: OBJETIVOS 33
CAPÍTULO IV: INSTRUMENTACIÓN, APARATOS Y REACTIVOS 37

4.1. Instrumentos y aparatos 39
4.1.1. Cromatógrafo de líquidos con detector de espectrometría
de masas (UPLC-MS) 39
4.1.2. Simulador solar 40
4.1.3. Balanza analítica 41
4.1.4. Sistema de concentración de muestras 41
4.1.5.pH-metro 42
4.2. Reactivos y disoluciones 43
4.2.1. Reactivos 43
4.2.2. Disoluciones 44
CAPITULO V: DESARROLLO Y VALIDACIÓN DEL MÉTODO DE SEPARACIÓN 45
5.1. Ensayos previos 47
5.2. Cromatografía líquida (LC)/ Espectrometría de masas (MS) 48
5.3. Condiciones cromatográficas 49
5.4. Patrón interno 52
5.5. Validación del método 53
5.5.1. Selectividad 53
5.5.2. Estudio de linealidad 56
5.5.3. Efecto matriz 58
5.5.4. Límites de detección (LD) y cuantificación (LC) 59
5.5.5. Precisión 59
CAPÍTULO VI: FOTODEGRADACÓN DE AMETOCTRADÍN 63
6. Fotodegradación de Ametoctradín en laboratorio 65

6.1. Condiciones experimentales 65
6.2. Metodología 66
6.3. Cinéticas de fotodegradación 67
6.4.1. Fotodegradación a pH ácidos 67
6.4.2. Fotodegradación a pH neutro 69
6.4.3. Fotodegradación a pH básico 71
6.4.4. Fotodegradación en disoluciones tampón 72
6.4.5. Fotodegradación en mosto 74
6.4. Evolución de Ametoctradín 75
6.5. Evolución de los productos de degradación 76
6.6.1. pH = 2,0 77
6.6.2. pH = 3,5 82
6.6. Determinación de residuos en el vino 86
6.7. Identificación de los productos de degradación 87
CAPÍTULO VII: CONCLUSIONES 93
CAPÍTULO VIII: BIBLIOGRAFÍA 97

CAPÍTULO I:
FUNGICIDAS EN VID


Fungicidas en vid Capítulo I
7
1.1. Enfermedades de la vid causadas por hongos
Las enfermedades más importantes en las que se centra la protección del
viñedo son el oídio, el mildiu y la podredumbre gris. Además pueden
aparecer otro tipo de hongos como la excoriosis, la eutipiosis y la yesca.
1.1.1. Oidio
El agente causal es el hongo Uncinula necator, originario de América del
Norte, pero ampliamente extendido en España. Este hongo ataca a todos
los órganos verdes de la vid, especialmente a los brotes, sarmientos y
racimos.
Entre los síntomas y daños más
destacados se encuentran la aparición
de un polvillo blanco-ceniza en las
hojas que puede llegar a cubrirlas por
completo, tal como se muestra en la
figura 1. Además tiene lugar la
formación de manchas blancas en los
brotes y sarmientos, las cuales van
creciendo y oscureciendo. Finalmente
el hongo pasa a los racimos y los seca.
El desarrollo del Oidio se ve favorecido por temperaturas elevadas,
atmósfera seca y noches frescas. Al tener un desarrollo externo se puede
combatir con una amplia variedad de productos y estrategias de control,
siendo importante la alternancia de los mismos para evitar resistencias.
Figura 1: Ataque de Oidio en hojas

Capítulo I Fungicidas en vid
8
1.1.2. Mildiu
Se trata de una de las enfermedades más conocidas y a su vez más
graves. Con condiciones ambientales favorables puede atacar a todos los
órganos verdes de la vid, provocando la pérdida de hasta el 50% o más de
la cosecha. Está provocada por el hongo Plasmopara vitícola y aparece
en regiones de clima cálido y húmedo durante el periodo de crecimiento
vegetativo.
Los síntomas más destacados son la aparición de “manchas de aceite” en
el haz de las hojas como se muestra en la figura 2 y de una pelusilla
blanquecina en el envés. Los ataques fuertes producen una desecación
parcial o total de las hojas que repercute en la cantidad y calidad de la
cosecha.
Figura 2: Mildiu en hojas y racimos
Otro de los síntomas son la curvatura de los brotes afectados y el
recubrimiento con una pelusilla blanca. También los racimos pueden sufrir
el ataque apareciendo una curvatura en S o adquiriendo un color pardo y
secándose como se puede observar en la figura 2. El mildiu sólo puede
prevenirse; la estrategia de protección consiste en tratar en el momento

Fungicidas en vid Capítulo I
9
oportuno para impedir o detener la germinación de las esporas. Para ello
se emplean tratamientos con cobre y productos orgánicos.
1.1.3. Botritis o podredumbre gris
El hongo Botrytis cinerea se manifiesta en los órganos herbáceos (hojas y
brotes) y ataca principalmente sobre los racimos próximos a la
maduración.
Los síntomas más importantes son la
aparición de amplias necrosis con aspecto
de quemaduras en las hojas. Los primeros
síntomas en brotes jóvenes y sarmientos se
caracterizan por la presencia de manchas
alargadas de color marrón recubiertas de
una pelusilla grisácea, oscureciéndose al
final de la maduración. Los ataques pueden
ocasionar la pérdida de algunos brotes
jóvenes, con la consiguiente disminución de la cosecha. Por otra parte los
granos presentan aspecto de podrido como se muestra en la figura 3.
A esta enfermedad también se le atribuyen otros aspectos como la
disminución de la calidad de los futuros vinos debido a la degradación de
las materias colorantes, la destrucción de la película que contiene las
sustancias aromáticas, la reducción del grado alcohólico, el aumento de
fijación de SO2 y la acidez volátil de los vinos.
La enfermedad se propaga por contacto y la lucha para combatirla no es
fácil ya que se trata de un hongo interno.
Figura 3: Granos enfermos de
Botritis

Capítulo I Fungicidas en vid
10
1.1.4. Excoriosis
Esta enfermedad está provocada por el hongo Phomopsis viticola Sacc,
pudiendo afectar a todos los órganos verdes de la vid, especialmente a los
sarmientos.
Sus síntomas se manifiestan como
puntuaciones o placas negras como las
que se observan en la figura 4, que
después se resquebrajan. Por otro lado,
las hojas también pueden sufrir el
ataque, presentando manchas oscuras.
La germinación de las esporas tiene
lugar exclusivamente en condiciones
húmedas, por tanto su desarrollo dependerá de la frecuencia de las lluvias.
Dentro de los métodos de control se recomienda quemar los restos de
poda y seguir un control químico durante el invierno.
1.1.5. Eutipiosis o eutipia
La eutipiosis es una enfermedad producida por el hongo Eutypa lata, cuyo
ataque se centra en el tronco y los brazos de las cepas, figura 5.
Es un hongo que penetra a través de los
cortes de poda. Los síntomas y daños más
destacados son la presencia de brotes
débiles y cortos, así como hojas más
pequeñas y deformadas, cloróticas y con
necrosis.
Las medidas más eficaces para erradicar
Figura 4: Ataque por Excoriosis
Figura 5: Ataque por Eutipiosis

Fungicidas en vid Capítulo I
11
esta enfermedad se basan en el tratamiento preventivo; consistente en la
poda y quema posterior de todos los sarmientos y brazos atacados, al igual
que todas las cepas muertas.
1.1.6. Yesca
Se trata de una enfermedad parasitaria producida por hongos (Stereum
hirsutum Per. y Phellinus igniarius Fr.) que penetran en la madera a través de
heridas importantes producidas en la poda y desarrollan el micelio en la
madera transformándola en yesca.
Los síntomas más importantes son la
desecación repentina de las cepas y la
aparición de una coloración parda bajo la
corteza de los brazos y los troncos. Al realizar
un corte del tronco se puede apreciar en el
centro madera amarilla, careada (yesca),
rodeada por una zona de madera
oscurecida y un anillo de madera sana de
espesor variable.
Los tratamientos para el control de esta enfermedad son difíciles. Las
medidas recomendadas consisten en la desinfección de las herramientas
de poda, podar en último lugar las cepas afectadas, quemar los restos de
poda y usar un producto protector.
1.2. Productos fitosanitarios
Según la Organización mundial de la salud (OMS), se define el producto
fitosanitario como aquella sustancia o mezcla de sustancias cuyo fin es
Figura 6: Presencia de Yesca

Capítulo I Fungicidas en vid
12
prevenir o destruir la acción de insectos, ácaros, moluscos, roedores,
hongos, malas hierbas, bacterias y otras formas de vida animal o vegetal
perjudiciales para la salud pública y para la agricultura durante la
producción, almacenamiento, transporte, distribución y elaboración de
productos agrícolas.
El término plaguicida es aplicado a las sustancias u organismos capaces
de exterminar toda vida animal o vegetal que pueda afectar a la salud, a
la alimentación o a la economía del hombre.
Dentro de los plaguicidas podemos encontrar diferentes tipos, entre ellos
destacan los insecticidas, acaricidas, molusquicidas, rodenticidas,
herbicidas y fungicidas. Estos últimos se emplean para la prevención y el
tratamiento de enfermedades causadas por hongos en diversos cultivos,
entre ellos la vid.
El empleo de plaguicidas ha supuesto beneficios para la economía
agrícola gracias a la eficacia y rapidez de acción ante plagas que
afectan al nivel de producción. Sin embargo estos productos también
traen consecuencias negativas ya que presentan problemas de toxicidad
para las personas y contaminación del medio ambiente.
Actualmente existe una multitud de productos que han sido retirados del
mercado debido a su peligrosidad, siendo sustituidos por otros con mejores
perfiles toxicológicos y ecotoxicológicos. Para ello se han llevado a cabo
diversos estudios sobre la persistencia y facilidad de eliminación de dichos
productos aplicados en campo, así como el estudio de los diferentes
productos de degradación formados a partir de ellos. En algunos casos
estos productos de degradación pueden presentar una toxicidad mayor
que los productos de partida.

Fungicidas en vid Capítulo I
13
Una vez aplicados en campo, estos productos pueden sufrir diversos
procesos de acumulación y/o dispersión generando graves problemas
medioambientales y sociales. También pueden quedarse retenidos en los
cultivos tratados generando residuos que afectaran a las características de
los productos agrícolas.
1.3. Sistema de producción integrada
El Sistema de producción integrada (SPI), se define como un nuevo sistema
de producción agraria que incluye diferentes técnicas y normas para el
tratamiento agrícola, reduciendo el consumo de fertilizantes químicos,
insecticidas, etc. El empleo de los mismos se lleva acabo solo cuando es
estrictamente necesario y siempre controlado por un técnico cualificado.
Se trata por tanto de un nuevo tipo de agricultura entre la convencional y
la ecológica cuya finalidad es aprovechar al máximo los recursos, así
como los mecanismos de producción naturales utilizando prácticas
compatibles con el medio ambiente. Este sistema asegura a largo plazo
una economía sostenible.
En cuanto a los beneficios de la producción integrada, se pueden distinguir
diferentes aspectos, en los que tanto el consumidor y los agricultores, como
el medio ambiente y la economía resultan beneficiados. Entre estos
beneficios destacan:
Mayor calidad y seguridad alimentaria para los consumidores.
Menor coste de producción y mayor rentabilidad para los
agricultores.

Capítulo I Fungicidas en vid
14
Reducción de la contaminación, empleando métodos menos
agresivos que respeten tanto la flora y la fauna como los recursos
naturales.
Productos diferenciados y con un valor añadido.
La filosofía de la Producción Integrada deriva de las directrices emanadas
de la Organización Internacional para la Lucha Biológica (OILB).
En el marco de la definición de Producción Integrada de la OILB, la
Producción Integrada de Uva se define como la producción económica
de uva de alta calidad, para cuya obtención se da prioridad a los
métodos ecológicamente más seguros y se minimizan la utilización de
agroquímicos y sus efectos secundarios negativos, para aumentar la
protección del medio ambiente y de la salud humana.
En la comunidad autónoma de La Rioja, la ordenación de la Producción
Integrada se inició a finales del año 2001 con la aprobación de su
reglamento y la posterior regulación del sistema en cuanto a controles,
autorizaciones de uso, registros y entidades de certificación.
Desde ese momento, se han ido publicando los reglamentos específicos
de cada cultivo, estableciéndose una serie de prácticas de cultivo
obligatorias, recomendadas y prohibidas. Dichas prácticas afectan a todas
las fases del proceso de producción, desde la implantación del cultivo
hasta su recolección y almacenamiento o elaboración y envasado.
1.4. Materia activa estudiada: Ametoctradín
1.4.1. Características físico-químicas
Ametoctradin es un fungicida de nueva generación desarrollado por BASF
que actúa como inhibidor de la respiración mitocondrial de los
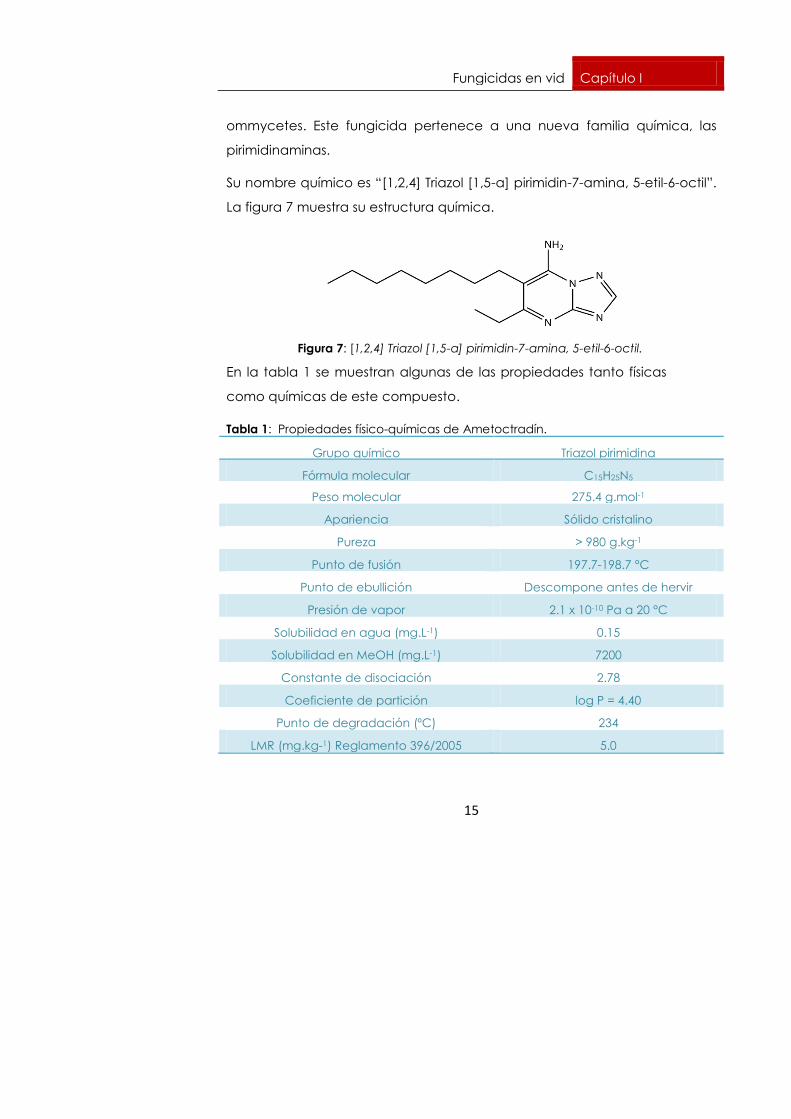
Fungicidas en vid Capítulo I
15
ommycetes. Este fungicida pertenece a una nueva familia química, las
pirimidinaminas.
Su nombre químico es “[1,2,4] Triazol [1,5-a] pirimidin-7-amina, 5-etil-6-octil”.
La figura 7 muestra su estructura química.
Figura 7: [1,2,4] Triazol [1,5-a] pirimidin-7-amina, 5-etil-6-octil.
En la tabla 1 se muestran algunas de las propiedades tanto físicas
como químicas de este compuesto.
Tabla 1: Propiedades físico-químicas de Ametoctradín.
Grupo químico Triazol pirimidina
Fórmula molecular C15H25N5
Peso molecular 275.4 g.mol-1
Apariencia Sólido cristalino
Pureza > 980 g.kg-1
Punto de fusión 197.7-198.7 °C
Punto de ebullición Descompone antes de hervir
Presión de vapor 2.1 x 10-10 Pa a 20 °C
Solubilidad en agua (mg.L-1) 0.15
Solubilidad en MeOH (mg.L-1) 7200
Constante de disociación 2.78
Coeficiente de partición log P = 4.40
Punto de degradación (ºC) 234
LMR (mg.kg-1) Reglamento 396/2005 5.0

Capítulo I Fungicidas en vid
16
Diferentes ensayos han demostrando que se trata de un fungicida con alta
selectividad y eficacia en cuanto a aplicaciones de pulverización
preventiva contra el mildiu en una amplia gama de cultivos de
especialidad.
(Merkel et al. 2012) estudiaron la eficacia de Ametoctradín para el control
de Plasmopara vitícola en uvas. Para ello llevaron a cabo la pulverización
del fungicida en uvas cultivados en campo. A las 72 horas del tratamiento
se tomó muestra, se lavó con agua de lluvia y se estudió la concentración
del fungicida en la capa cerosa tratada así como en el agua de lavado.
Los resultados reflejaron alta afinidad por la capa cerosa epicuticular y
resistencia a la lluvia.
También realizaron una pulverización con Orvego (fungicida compuesto
de Ametoctradín y dimetomorfo) sobre plantas de patata. Posteriormente
se rociaron con lluvia simulada y se estudió la persistencia del compuesto
en la zona epicuticular de la epidermis de la hoja. Los resultados obtenidos
indicaron la formación de una película estable protectora contra el
ataque de hongos.
Por último llevaron a cabo un estudio en diferentes países sobre la
pulverización de Orvego en campos de tomate, pepino y melón,
demostrando su eficacia contra el tizón tardío y mildiu.
1.4.2. Perfil toxicológico y ecotoxicológico
Este fungicida presenta un excelente perfil toxicológico. Según estudios
realizados en mamíferos se ha demostrado que Ametoctradín no es
perjudicial para la salud de los mismos.

Fungicidas en vid Capítulo I
17
Por otro lado también presenta un excelente perfil ecotoxicológico, siendo
su uso muy adecuado en cultivos integrados en programas de gestión.
Prácticamente no presenta toxicidad para las aves, mamíferos, abejas
lombrices de tierra y otros macroorganismos del suelo. Además el uso de
Ametoctradín de acuerdo con las buenas prácticas agrícolas no
representa ningún riesgo para los ecosistemas acuáticos.
1.4.3. Modo de acción
Ametoctradín es un potente inhibidor de la cadena respiratoria
mitocondrial en patógenos diana. Más específicamente, interfiere con el
complejo III, también llamado complejo BC1. El complejo III es una
membrana del complejo proteico, que consta de 11 subunidades de la
cadena respiratoria.
Al inhibir el complejo III, Ametoctradín perjudica el transporte de
electrones en la cadena respiratoria del patógeno, por lo que es incapaz
de generar la energía requerida para mantener el organismo vivo. En la
figura 8 se muestra el sistema de transporte electrónico en la mitocondria
de los hongos.
La investigación ha demostrado que este fungicida no presenta resistencia
cruzada a fenilamidas (por ejemplo, metalaxil), Qo inhibidores (por
ejemplo estrobilurinas) ni a amidas de ácidos carboxílicos (por ejemplo
dimetomorfo).

Capítulo I Fungicidas en vid
18
Figura 8: Sistema de transporte electrónico en la mitocondria de los hongos.
1.4.4. Caracterización biológica
Macroscópicamente, Ametoctradín es un inhibidor muy eficaz de la
formación, liberación, motilidad y germinación de zoosporas (órganos
contaminantes del hongo). En concentraciones muy bajas, conduce
rápidamente a la ruptura de zoosporas, poniendo fin de inmediato
al desarrollo de las etapas infecciosas del patógeno. La figura 9 muestra las
diferencias entre una zoospora sin tratamiento y otra tratada con
Ametoctradín.
A) B)
Figura 9: A) Zoospora sin tratamiento y B) ruptura de zoospora después del
tratamiento con Ametocradín.

Fungicidas en vid Capítulo I
19
Ametoctradín presenta alta afinidad por la capa cerosa de la epidermis
de la hoja y mediante adsorción forma una película protectora contra el
ataque de los hongos.
Por otro lado presenta muy buena resistencia a la lluvia, sin embargo, bajo
la influencia de la humedad, una pequeña pero efectiva porción del
ingrediente activo se distribuye gradualmente a través de la película
protectora conduciendo a un aumento de la protección.
Finalmente menos del 10% del ingrediente activo aplicado es absorbido
por las hojas después de 1-7 días. La mayor parte del producto permanece
en su superficie.
Dado que no presenta resistencia cruzada a otras clases de fungicidas, se
han realizado formulaciones listas para usar en combinación con otros
ingredientes activos que presentan un modo de acción diferente. Esto
garantiza su eficacia a largo plazo.
Por otro lado el mayor rendimiento se obtiene aplicándolo como un spray
protector antes de que la enfermedad se establezca en el cultivo, ya que
actúa de manera preventiva y no erradicante o curativa

CAPÍTULO II:
DEGRADACIÓN DE FUNGICIDAS


Degradación de fungicidas Capítulo II
23
2.1. Mecanismos de degradación
En la figura 10 se muestran los procesos por los cuales pueden estar
afectados los fungicidas una vez liberados al medio ambiente. Se trata de
procesos químicos, físicos, biológicos o fotoquímicos (Kot-Wasik et al. 2004).
Figura 10: Cambios de los fungicidas en el medio ambiente
Por un lado los procesos físicos conllevan el transporte de los fungicidas
(volatilización, dispersión, sedimentación, mezcla y dispersión en suelo y
sedimentos) determinando su distribución temporal y espacial en el
entorno medioambiental.
Por otro lado, los procesos químicos, biológicos y fotoquímicos pueden
producir la transformación mediante hidrólisis, biodegradación o fotólisis,
resultando de gran importancia para la determinación de la persistencia
del compuesto (Gavrilescu et al. 2005).
Estos procesos son el principal mecanismo de eliminación de fungicidas del
medio ambiente. En ellos intervienen factores como la reactividad del

Capítulo II Degradación de fungicidas
24
compuesto así como parámetros medioambientales tales como la
temperatura, la intensidad de la luz y la composición bacteriana.
Un aspecto importante de estos procesos es la formación de productos de
degradación a partir de los compuestos iniciales, los cuales pueden
presentar una toxicidad mayor tanto para el hombre como para el medio
ambiente (Kot-Wasik et al. 2006).
Por este motivo es importante estudiar el comportamiento de los fungicidas
en el medioambiente, su persistencia (resistencia a cualquier tipo de
degradación), los productos de degradación así como su toxicidad.
2.2. Cinética de los procesos de degradación
Para determinar las cinéticas de degradación se representa la
concentración frente al tiempo y se ajusta a la curva de regresión que
ofrezca un mayor valor del coeficiente de correlación, R2.
Aunque la degradación de los pesticidas puede seguir cinéticas de
distintos órdenes, en general, pueden describirse como reacciones de
primer orden (Kot-Wasik et al. 2004).
La ecuación 1 expresa un ajuste de primer orden:
kcdt
dc1 Ecuación
Siendo c la concentración (mg.L-1) de compuesto a degradar, t (s) el
tiempo de exposición al proceso degradativo y k (s-1) la constante de
velocidad de primer orden.
La ecuación 2 se obtiene por integración de la ecuación 1 en el tiempo:
ktectc 0)(2Ecuación

Degradación de fungicidas Capítulo II
25
Siendo c0 la concentración inicial (mg.L-1) del compuesto para un tiempo
t= 0 y ct la concentración (mg.L-1) al tiempo t.
La concentración de compuesto es una función exponencial del tiempo
de degradación, tal y como puede observarse en la ecuación 2. En estos
casos, la representación del logaritmo de las concentraciones en función
del tiempo conlleva a una curva de regresión lineal cuya pendiente se
puede relacionar con la constante de degradación (k).
Para aquellos casos en los que se cumple la cinética de primer orden se
calcula el tiempo de reducción a la mitad t1/2 que corresponde al tiempo
que tarda la concentración inicial de una molécula en reducirse a la
mitad. Así, a partir de la ecuación 3, se obtiene la ecuación
correspondiente, ecuación 4:
kt
2ln4Ecuación 2/1
2.3. Estudios de fotodegradación
La fotodegradación es la transformación fotoquímica de una molécula en
otras, normalmente de menor peso molecular. A su vez, las
transformaciones fotoquímicas son las reacciones químicas producidas por
la absorción de radiación ultravioleta-visible en la atmósfera, la superficie
del suelo o del agua. Dentro de estas transformaciones se pueden
destacar las fotocicloadiciones, fotodescarbonilaciones, fotodescarbo-
xilaciones, fotoisomerizaciones, fotosustituciones, fotoreducciones, etc
(IUPAC 2007).
ktcct )ln()ln(3Ecuación 0

Capítulo II Degradación de fungicidas
26
La fotodegradación tiene lugar cuando el espectro de radiación UV/VIS
del fungicida solapa con el espectro de radiación de la luz solar. Como
consecuencia se puede producir la rotura de enlaces, siendo los grupos
-OH, -SH, C=O, -Cl, -N=, así como los dobles enlaces sobre todo si son
conjugados los más afectados. Antes de finalizar la fotodegradación se
forman productos intermedios, cuya identificación es de gran importancia
ya que pueden poseer una mayor toxicidad que los compuestos de los
que proceden.
En general, las reacciones fotoquímicas se clasifican en directas o
indirectas. En la fotólisis directa la reacción se produce por la absorción
directa de la luz por el propio fungicida, mientras que en la fotólisis
indirecta la luz es absorbida por otros compuestos fotosensibilizadores o
iniciadores de radicales, los cuales reaccionan con el propio fungicida
(Burrows 2002).
Un esquema de los mecanismos de fotodegradación se representa en la
figura 11:
Fotodegradación directa
Compuesto + hν → Compuesto*
Compuesto* → Fotoproductos
Fotodegradación indirecta
Sensibilizador o iniciador de radicales (S) + hν → S*
S* + Compuesto → Compuesto* + S ó
S* + Compuesto → Compuesto· + S·
Compuesto* → Fotoproductos
Compuesto· → Fotoproductos
Figura 11: Mecanismos de fotodegradación

Degradación de fungicidas Capítulo II
27
En el medio acuático, estos dos mecanismos ocurren simultáneamente
debido a la existencia de microorganismos, algas o sustancias húmicas,
que actúan de fotosensibilizadores acelerando la fotodegradación de los
compuestos orgánicos presentes en el medio.
Diversos autores han realizado estudios en el laboratorio enfocados a la
identificación de los productos resultantes de los procesos de
fotodegradación y a estudios cinéticos.
Morrica et al. (2005) realizaron un estudio del fungicida cymoxanil en
disolución acuosa tampón pH= 5, para evitar procesos de hidrólisis. La
irradiación se llevó a cabo con lámpara de mercurio (20W), obteniéndose
una cinética de primer orden. El seguimiento cinético y la identificación de
los productos de degradación se realizaron mediante cromatografía
líquida de alta presión con detector ultravioleta (HPLC-UV) y cromatografía
líquida de alta presión acoplada a espectrometría de masas (HPLC-MS).
Además para la confirmación de los productos de degradación
encontrados se empleó un análisis masas-masas (MS-MS) previa
concentración de la muestra. El mecanismo de degradación propuesto
implicaba la formación de anillos bajo mecanismos radicalarios.
Chen et al. (2009) realizaron un estudio de fotodegradación de pirimicarb
en disolución acuosa. La irradiación se llevó a cabo con lámpara de
mercurio, obteniéndose una degradación total a los 180 minutos. Para el
análisis tanto del pirimicarb como de los productos de degradación se
realizó una inyección directa de las muestras previamente filtradas, en
cromatografía líquida con trampa iónica acoplada a espectrometría de
masas (LC-IT-MS). La identificación de los productos de degradación se
llevó a cabo mediante cromatografía líquida masas-masas (LC-MS-MS) y
estudio de RMN de 1H y 13C. De acuerdo a los metabolitos encontrados se
propuso un mecanismo de degradación que incluía rupturas homolíticas

Capítulo II Degradación de fungicidas
28
de los enlaces N-C y C-O, así como la oxidación del grupo N-metil unido
con el grupo dimetilamina del pirimicarb.
Krieger et al. (2000) realizaron un estudio sobre la fotodegradación del
herbicida florasulam, perteneciente a la familia triazol-pirimidina en aceite
y sistemas acuosos (disolución tampón y agua natural). La irradiación tuvo
lugar por luz solar natural durante 32 días, obteniéndose una cinética de
degradación de primer orden. El seguimiento de la degradación se realizó
mediante análisis por HPLC-UV. La cinética de degradación fue más rápida
en medios acuosos naturales debido a la importante contribución de
procesos fotolíticos indirectos.
A parte de los procesos fotolíticos, también han sido estudiados por
algunos autores los procesos fotocatalíticos. Dichos procesos presentan
condiciones similares a las de los procesos fotolíticos pero con presencia de
una sustancia que actúe como catalizador.
Agüera et al. (2009) llevaron a cabo el estudio fotocatalítico de pirimetanil
en disolución acuosa en presencia de TiO2 como catalizador. La irradiación
tuvo lugar mediante luz solar natural. La degradación siguió una cinética
de primer orden observándose degradación total a los 230 minutos de
irradiación. Se realizó un estudio cualitativo y cuantitativo de los productos
de degradación por cromatografía de gases masas (GC-MS) previo paso
de extracción en fase sólida (SPE) o extracción líquido-líquido (LLE) y por
filtración e inyección directa por cromatografía líquida con fuente de
ionización química a presión atmosférica acoplada a espectrometría de
masas (LC-API-MS). Para la identificación de los productos de degradación
se emplearon patrones comerciales de comparación y la librería comercial
Wiley 275. Una vez identificados los productos de degradación se
propusieron dos rutas metabólicas: ataque de los radicales hidroxilo al

Degradación de fungicidas Capítulo II
29
benceno o al anillo pirimidina con apertura del anillo e hidrólisis
fotoinducida de la molécula por los enlaces del grupo amina.
Calza et al. (2004) realizaron una fotodegradación de mepanipyrim en
disolución acuosa catalizada por TiO2. La irradiación se llevó a cabo con
una lámpara de arco de xenón, obteniéndose una cinética de primer
orden con una degradación total a los 15 minutos de irradiación. El análisis
directo de las muestras previamente filtradas se llevó a cabo en HPLC-MS.
Para la identificación y caracterización de los intermedios de reacción se
llevó a cabo un análisis de masas en tándem (MSn), proponiéndose varias
vías oxidativas y reductivas como rutas de degradación.
2.4. Análisis de residuos de fungicidas en muestras enológicas
Los tratamientos con productos fitosanitarios para controlar plagas y
enfermedades de los cultivos, dejan residuos de plaguicidas. Esto es
debido a que algunos de estos productos no se degradan de forma
natural, por lo que tanto ellos como sus productos de degradación
quedan en o sobre los productos cosechados y sus productos de
transformación como es el caso de la uva y el vino elaborado.
Los residuos de los plaguicidas aplicados en campo pueden pasar de las
uvas al mosto y finalmente al vino durante el proceso de vinificación
(Cabras et al. 2000). Este hecho puede tener como consecuencia la
alteración de la calidad organoléptica de los vinos, interferencias en el
proceso de fermentación y lo más importante, pueden resultar tóxicos para
el consumidor (Jiménez et al. 2007).
Hoy en día la existencia de residuos de pesticidas en uvas está regulado
por los LMR en uva, los cuales alcanzan bajos niveles de concentración

Capítulo II Degradación de fungicidas
30
(µg.mL-1), esperándose detectar valores significativamente inferiores en
vino (Flamini et al. 2006).
Dado que actualmente no existen LMR establecidos en vino, a excepción
de algunos compuestos, uno de los objetivos perseguidos por la química
analítica es el desarrollo de métodos de análisis que permitan obtener
resultados precisos y exactos de análisis de residuos en esta matriz. De esta
forma se podrán establecer y controlar dichos LMR.
En la revisión bibliográfica se han encontrado diferentes técnicas para la
extracción y concentración de pesticidas en muestras de uva, mosto y
vino. Entre estas técnicas se encuentra la extracción líquido-líquido (LLE)
con diclorometano empleada por (Vaquero et al. 2008a) para el análisis
de cyprodinil y fludioconil en muestras de mosto y vino así como en
muestras durante la fermentación alcohólica. (Vaquero et al. 2008b)
también emplearon (LLE) con n-Hexano para el estudio de pirimetanil,
metalaxil, diclorofluanid y penconazol en muestras sintéticas y reales de
mosto.
La extracción en fase sólida (SPE) con cartuchos Oasis HLB y metanol como
disolvente de elución fue empleada por (Economou et al. 2009) para la
determinación de 46 fungicidas de diferente familia química y sus
productos de degradación en vino. Otro estudio realizado por (Vaquero et
al. 2009a) consistió en el análisis simultáneo de pirimetanil, penconazol,
metalaxil y penconazol en mosto y vino mediante (SPE) con cartuchos de
octadecilsilano junto con acetato de etilo como disolvente de elución.
También (Vaquero et al. 2009b) empleó esta técnica para el estudió del
porcentaje de absorción de dichos fungicidas en uvas, así como su
distribución entre la superficie, la piel y la pulpa.

Degradación de fungicidas Capítulo II
31
La microextracción en fase sólida (SPME) acoplada a cromatografía
líquida de alta presión con detector diodo array (SPME-HPLC-DAD) fue
empleada por (Millan et al. 2003) para el estudio de 6 fungicidas
organoclorados en vino.
Otra técnica de extracción empleada por (Lagunas et al. 2011) fue la
extracción asistida por microondas (MAE) para el análisis de 8 pesticidas en
uvas. Las condiciones óptimas fueron 2 gramos de uva con 10 mL de
hexano-acetona (1:1 v/v) a 105ºC durante 10 minutos con una potencia
de microondas de 600 W.
Además de estas técnicas, también existen otras que han tenido éxito en el
pre-tratamiento de residuos de pesticidas en muestras líquidas. Entre ellas
encontramos la extracción por barra agitadora (SBSE) en combinación con
desorción térmica-capilar GC-MS (TD-CG-MS) empleada por (Sandra et al.
2001) para analizar fungicidas de la familia dicarboximida en vino. También
(Viñas et al. 2008) emplearon (SPME) acoplada a cromatografía líquida
ultra resolución (UPLC) para la determinación del fungicida oxalazol en
mosto y vino
Para la identificación y cuantificación de pesticidas en mosto y en vino se
emplea la cromatografía líquida (LC) o la cromatografía de gases (GC)
acoplada a diferentes detectores.
Entre las técnicas de LC se encuentra la cromatografía líquida acoplada a
espectrometría de masas con tiempo de vuelo y fuente de ionización de
electrospray empleada por (Fontana et al. 2011) para la determinación de
15 fungicidas en vino, la cromatografía líquida de alta resolución con
detector diodo array (HPLC-DAD) utilizada por (Lagunas et al. 2012) para el
estudio cinético de la fotocatálisis de piraclostrobín mientras que para el
estudio de productos de degradación se empleó la cromatografía líquida

Capítulo II Degradación de fungicidas
32
acoplada a espectrometría de masas (LC-MS/MS). Por otra parte (Viñas et
al. 2008) realizaron la determinación de oxazol en vino y mosto mediante
cromatografía líquida ultra resolución (UPLC) con diodo array.
Dentro de GC, la cromatografía de gases acoplada a espectrometría de
masas con trampa iónica (GC-ITMS) fue utilizada por (Angioni et al. 2005)
para la determinación del fungicida zoxamida en uva, mosto y vino y la
cromatografía de gases con detector NPD (GC-NPD) por (Vaquero et al.
2009b) para la determinación de los residuos de 4 fungicidas en uva.
También llevaron a cabo una confirmación adicional por cromatografía
de gasas acoplada a espectrometría de masas (GC-MS). Otro estudio
realizado con GC fue llevado a cabo por (Likas et al. 2007) para la
determinación de residuos de famaxadona, trifloxistrobin y fenhexamid en
uvas y vino. Para ello emplearon 3 tipos de detectores: nitrógeno-fósforo
(NPD), de captura electrónica (ECD) y espectrometría de masas con
trampa iónica.

CAPÍTULO III:
OBJETIVOS


Objetivos Capítulo III
35
3. Objetivos
El objetivo principal de este trabajo ha sido estudiar el proceso de
fotodegradación “in vitro” de una nueva materia activa fúngica,
Ametoctradín. Para lograrlo se han fijado objetivos más concretos:
Desarrollar el método de separación adecuado para la
determinación de residuos del fungicida Ametoctradín mediante
cromatografía líquida acoplada a espectrometría de masas.
Validar el método desarrollado.
Estudiar la fotodegradación del fungicida en condiciones de
laboratorio (“in vitro”).
Aplicar el método analítico desarrollado para la detección y
cuantificación de los residuos del fungicida y de los productos de
degradación obtenidos en el proceso degradativo.
Estudiar las cinéticas de reacción en las condiciones estudiadas,
evaluando la influencia del medio en los procesos de degradación.
Hacer un estudio de la evolución del pesticida y sus productos de
degradación a lo largo del proceso de fotodegradación.
Estudiar la disipación de Ametoctradín en bodega durante la
elaboración del vino y determinar los residuos de Ametoctradín en
el vino final.
Identificación de los productos de degradación obtenidos.

CAPÍTULO IV:
INSTRUMENTACIÓN, APARATOS Y
REACTIVOS


Instrumentación, aparatos y reactivos Capítulo IV
39
4.1. Instrumentos y aparatos
Para llevar a cabo este trabajo de investigación se emplearon los
siguientes instrumentos y aparatos:
4.1.1. Cromatógrafo de líquidos con detector de espectrometría de masas
(UPLC/MS)
El UPLC-MS que se utilizó fue un Bruker MicroTOF-Q, como el que muestra la
figura 12, equipado con los siguientes elementos:
Una columna ACQUALITY UPLC BEH C18 (2,1 x 100 mm; 1,7 µm).
Detector de espectrometría de masas con detección por tiempo
de vuelo.
Fuente de ionización de electrospray.
El software empleado para el tratamiento de los datos extraídos de los
análisis fue Bruker Daltonics DataAnalysis.
Figura 12: Equipo UPLC-MS

Capítulo IV Instrumentación, aparatos y reactivos
40
4.1.2. Simulador solar
Para el estudio de las fotodegradaciones en el laboratorio se empleó un
simulador solar Orion, compuesto de una fuente de alimentación y una
lámpara de arco de xenón, tal como se muestra en la figura 13.
La muestra a degradar se depositó en una cubeta de cuarzo de 25 ml de
capacidad de 10,5 cm de longitud y 2,5 cm de diámetro, la cual se puede
ver en la figura 14, a 5 cm de distancia con la fuente de radiación.
Para evitar cualquier incidencia de la luz externa sobre la muestra, se aisló
la celda con papel de aluminio y la degradación se llevó a cabo en un
cuarto acondicionado para trabajar en ausencia total de luz.
Figura 13: Equipo solar
Figura 14: Celda de cuarzo donde se introdujo la muestra a degradar.

Instrumentación, aparatos y reactivos Capítulo IV
41
4.1.3. Balanza analítica
En este trabajo se empleó una balanza digital Sartorius BL 120S (con una
precisión de la décima de miligramo) como la que aparece en la figura
15, para pesar todos los patrones y reactivos empleados en el desarrollo
experimental.
Figura 15: Balanza digital Sartorius BL 120S
4.1.4. Sistema de concentración de muestras
Para el estudio de los productos de degradación se llevó a cabo la
concentración de las muestras mediante un sistema de vacio Büchi
Rotavapor R-200, equipado con un baño Büchi Heating Bath B-490 a 30ªC,
como se muestra en la figura 16.

Capítulo IV Instrumentación, aparatos y reactivos
42
Figura 16: Büchi Rotavapor R-200, con un baño Büchi Heating Bath B-490.
4.1.5. pH-Metro
Para ajustar el pH de las disoluciones se empleó un pH metro “pH meter
GLP 21” CRISON, como el que se muestra en la figura 17, así como para
controlar la variación de pH a lo largo del proceso degradativo.
Figura 17: pH meter GLP 21 CRISON

Instrumentación, aparatos y reactivos Capítulo IV
43
4.2. Reactivos y disoluciones
4.2.1. Reactivos
Para la realización de este estudio la materia activa Ametoctradín, fue
suministrada por la casa comercial Dr. Ehrenstorfer con una pureza del
99,8% (m/m).
Metribucina fue la materia activa utilizada como patrón interno,
suministrada por la casa comercial Riedel-de Haën con una pureza del
99,8% (m/m).
En la tabla 2 se recogen los reactivos y disolventes utilizados con su grado
de pureza y la casa comercial que los suministra:
Tabla 2: Reactivos y disolventes empleados.
Reactivo/Disolvente Grado de pureza Casa comercial
Agua desionizada Grado Milli Q Millipore
Metanol Grado HPLC Scharlab
Ácido fórmico Grado MS Sigma-Aldrich
Ácido clorhídrico 37% Grado ACS, ISO Scharlau
KH2PO4 98 - 100,5% Panreac
Na2HPO4· 2H2O 99 – 101% Panreac
Ftalato ácido de
potásico 99,95 – 100,05 % Panreac
Hidróxido de sodio 98 % Prolabo

Capítulo IV Instrumentación, aparatos y reactivos
44
4.2.2. Disoluciones
La disolución patrón concentrada de Ametoctradín se preparó en metanol
en una concentración de 500 mg.L-1 por pesada directa de la materia
activa. A partir de esta se prepararon disoluciones de 100 mg.L-1 y las
correspondientes disoluciones de trabajo.
La disolución concentrada de patrón interno, Metribucina, se preparó en
metanol en una concentración de 1000 mg.L-1 por pesada directa de la
materia activa patrón. A partir de ésta se prepararon las disoluciones
intermedias de 100, 10 y de 1 mg.L-1.
Todas las disoluciones se mantuvieron a una temperatura de -20 ºC durante
un periodo máximo de 3 meses.
Las disoluciones tampón utilizadas en el estudio se prepararon de la
siguiente manera:
Disolución tampón pH 3,5: 1,0211 g de ftalato ácido de potasio
disuelto en 50 mL de agua MilliQ + 8,2 mL de ácido clorhídrico 0,1 M
en un volumen total de 100 mL.
Disolución tampón pH 7,0: 75 mL de disolución A + 25 mL de
disolución B. Disolución A: 0,9073 g de KH2PO4 disuelto en 100 mL de
agua MilliQ. Disolución B: 1,1870 g de Na2HPO4· 2H2O disuelto en 100
mL de agua MilliQ.
Por otra parte las disoluciones ácidas y la disolución básica se
prepararon como se indica a continuación:
Disoluciones ácidas pH 2,0 y 3,5: disoluciones acuosas acidificadas
añadiendo gota a gota una disolución de ácido clorhídrico 0,1 M.
Disolución básica pH 11,0: disolución acuosa basificada añadiendo
gota a gota una disolución de hidróxido de sodio 0,1 M.

CAPÍTULO V:
DESARROLLO Y VALIDACIÓN DEL
MÉTODO DE SEPARACIÓN


Desarrollo y validación del método de separación Capítulo V
47
5.1. Ensayos previos
Para analizar el resultado de las reacciones de degradación, las cuales
puedan contener mezcla del fungicida de partida junto con sus productos
de degradación se ha llevado a cabo un proceso de separación e
identificación que permite separar los compuestos y confirmar la presencia
de los mismos en las muestras a analizar.
En el desarrollo de este trabajo, se ha perseguido el objetivo de determinar
los compuestos directamente de la matriz, evitando así los pasos
intermedios de tratamiento de muestra. De esta manera, se ha obtenido la
ventaja de reducir el consumo de disolventes y evitar las pérdidas de
compuesto que conllevan los pasos intermedios.
Debido a la baja solubilidad del fungicida en disolución acuosa se tuvieron
que realizar una serie de pruebas de solubilidad en disoluciones
hidroalcohólicas con distintas relaciones de metanol-agua. En ellas se
analizaba la repetibilidad de las muestras acuosas añadiendo metanol en
un rango del 10%(v/v) al 50%(v/v). Según estos ensayos se estableció que el
mínimo porcentaje necesario para la completa disolución del compuesto
fue del 50% (v/v).
Actualmente, no se han encontrado referencias sobre la determinación e
identificación de este compuesto.

Capítulo V Desarrollo y validación del método de separación
48
5.2. Cromatografía líquida (LC)/espectrometría de masas (MS)
La LC-MS es considerada una técnica muy atractiva para el análisis de
residuos de pesticidas debido a la eficiente separación, identificación y
cuantificación de analitos,.
Además mediante la espectrometría de masas es posible llevar a cabo la
confirmación y cuantificación de compuestos dentro de un amplio
espectro de plaguicidas.
Actualmente se dispone de técnicas de interfase de presión atmosférica,
las cuales combinan alta sensibilidad y selectividad con cuantificación
fiable por lo que la LC-MS ha experimentado un gran avance.
Hasta mediados de los 90 la mayoría de aplicaciones en análisis de
residuos de pesticidas involucraban interfases de termospray (TSP) o de haz
de partículas (PB). A pesar de exitosas aplicaciones, esta técnica aun no
estaba bien adaptada en la práctica regular debido a su alto coste y a
variaciones significativas en la sensibilidad.
Sobre mediados de los 90, gracias al desarrollo y disponibilidad de
interfases de ionización a presión atmosférica (API), la mayoría de esos
problemas fueron reducidos o eliminados. Generalmente en API se
distingue entre ionización electrospray (ESI) o ionización química a presión
atmosférica (APCI). En este trabajo se empleó la ESI.
La técnica API aplicada en los métodos de LC-MS, es una técnica de
ionización suave que predominantemente produce la protonación [M+H]+
o la desprotonación [M-H]- molecular de los iones en modo de ionización
positiva (PI) o en negativa (NI) respectivamente (Hogendoorn et al. 2000).

Desarrollo y validación del método de separación Capítulo V
49
5.3. Condiciones cromatográficas (LC-MS)
En este trabajo el equipo instrumental empleado fue un Bruker MicroTOF-Q,
siendo las condiciones cromatográficas las siguientes:
La fase móvil fue constituida por una mezcla de disolvente A y B:
o Disolvente A: Metanol
o Disolvente B: mezcla de H2O + 0,1 % Ácido fórmico
La tabla 3 muestra el gradiente utilizado durante el tiempo de análisis.
Tabla 3: Gradiente de los disolventes empleados en LC-MS
Tiempo (min) % A % B
Inicial 10 90
1,00 10 90
1,50 70 30
2,00 70 30
2,70 70 30
3,00 100 0
5,90 100 0
6,00 10 90
La velocidad de flujo de la fase móvil fue de 0,45 mL min-1.
La fuente de ionización, como ya se ha dicho anteriormente fue de
electrospray, en modo de ionización positiva.
La columna cromatográfica ACQUALITY UPLC BEH C18 (2,1 x 100
mm; 1,7 µm) y la temperatura de dicha columna fue de 40ºC.
El volumen de muestra inyectado fue, en todos los casos, de 1 µL.

Capítulo V Desarrollo y validación del método de separación
50
La temperatura de la muestra fue de 10 ºC.
La cuantificación se realizó por el método de patrón interno
utilizando para ello Metribucina.
En estas condiciones cromatográficas seleccionadas los tiempos de
retención así como las relaciones m/z de los iones son mostrados en la
tabla 4:
Tabla 4: Tiempos de retención e iones de los analitos en LC-MS
Compuesto Tiempo (min) Ion (m/z)
Metribucina 2,30 215,09
Ametoctradín 3,60 276,22
En la figura 18 se muestra un cromatograma con los picos correspondientes
al fungicida estudiado y al patrón interno. En las figuras 19 y 20 se muestran
respectivamente sus espectros de masas.

Desarrollo y validación del método de separación Capítulo V
51
Figura 18: Cromatograma con la identificación de los compuestos (0.1 mg.L-1)
1) Metribucina (P.I.) y 2)Ametoctradín. Disolvente: 50% (v/v) MeOH: H2O.
Figura 19: Cromatograma de masas para Metribucina
215.1058
573.2790
1. +MS, 2.24-2.35min #(266-280), -Peak Bkgrnd
0
250
500
750
1000
1250
1500
Intens.
200 400 600 800 1000 1200 1400 m/z
1
2

Capítulo V Desarrollo y validación del método de separación
52
Figura 20: Cromatograma de masas para Ametoctradín
5.4. Patrón interno
El método de patrón interno es ampliamente utilizado para la
cuantificación en espectrometría de masas. Este método es especialmente
útil cuando la cantidad de muestra introducida y la respuesta del aparato
pueden variar de un “run” a otro (Dass 2007)
En la realización de este método se añadió la misma concentración de
patrón interno (0,1 mg.L-1) a todas las muestras objeto de análisis. Como
parámetro analítico se empleó la relación entre el área de pico del analito
y la del patrón interno.
El compuesto elegido como patrón interno fue Metribucina, un herbicida
perteneciente a la familia triazinona, cuya estructura se muestra en la
figura 21.
276.2312
402.3763 573.4386
2. +MS, 3.53-3.64min #(420-433), -Peak Bkgrnd
0.0
0.2
0.4
0.6
0.8
1.0
4x10
Intens.
200 400 600 800 1000 1200 1400 m/z

Desarrollo y validación del método de separación Capítulo V
53
Este compuesto tiene la ventaja de ser una sustancia de características
similares a las del analito, no se encuentra presente en las muestras (no se
emplea en el tratamiento de campo en vid), y posee unos grupos
funcionales idóneos para la ionización por electrospray.
Además su pico cromatográfico no presenta solapamiento con el del
analito objeto de estudio.
Figura 21: 4-amino-6-tert-butil-4,5-dihidro-3-metiltio-1,2,4-triazin-5-ona
5.5. Validación del método
La validación del método implica realizar un estudio de la selectividad, la
linealidad, el efecto matriz, los límites de detección y cuantificación y la
precisión (repetibilidad y reproducibilidad).
5.5.1. Selectividad
La selectividad expresa la capacidad del método para medir con
exactitud el analito en presencia de otros compuestos que puedan formar
parte de la matriz, sin producir interferencias.
Se mide comparando la matriz sin analito con la matriz fortificada. Para ello
se analizaron muestras de mosto y vino fortificados y sin fortificar.
Capítulo V Desarrollo y validación del método de separación
54
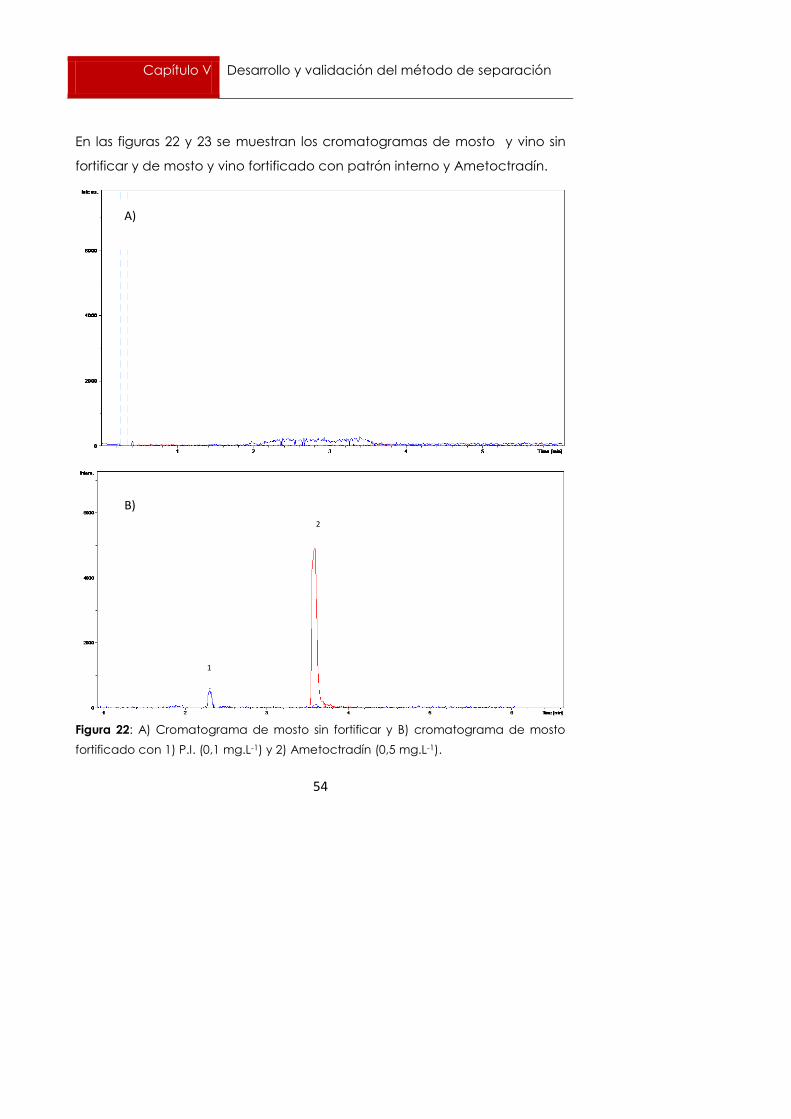
En las figuras 22 y 23 se muestran los cromatogramas de mosto y vino sin
fortificar y de mosto y vino fortificado con patrón interno y Ametoctradín.
Figura 22: A) Cromatograma de mosto sin fortificar y B) cromatograma de mosto
fortificado con 1) P.I. (0,1 mg.L-1) y 2) Ametoctradín (0,5 mg.L-1).
A)
B)
1
2

Desarrollo y validación del método de separación Capítulo V
55
Figura 23: A) Cromatograma de vino sin fortificar y B) cromatograma de vino
fortificado con 1)P.I. (0,1 mg.L-1) y 2) Ametoctradín (0,5 mg.L-1).
Como se puede observar, ni en mosto ni en vino aparece ningún pico con
la misma masa ni el mismo tiempo de retención que el patrón interno ni
Ametoctradín.
A)
B)
1
2

Capítulo V Desarrollo y validación del método de separación
56
Por tanto se trata de un método selectivo ya que en la matriz no
aparecieron señales cromatográficas que pudieran interferir en las señales
de los analitos.
5.5.2 Estudio de la linealidad
La linealidad es la capacidad del método analítico para obtener
resultados directamente proporcionales a la concentración o cantidad de
analito en un rango definido; es decir, para establecer una relación
directamente proporcional entre el resultado y la concentración del
analito en la muestra. Según la ecuación 5:
B + cA =relativa Área 5Ecuación
Siendo A la pendiente, B la ordenada en el origen y c la concentración en
mg.L-1.
Para construir la curva de calibrado se representó el área relativa del
analito (cociente entre el área absoluta del analito y el área absoluta del
patrón interno) frente a la concentración del analito. La representación
tuvo lugar a 7 niveles de fortificación (0.05, 0.1, 0.5, 1, 1.5, 3 y 5 mg.L-1) por
triplicado y con inyección por duplicado en LC-MS. La calibración se llevó
a cabo en 3 matrices: disolvente, en este caso agua, mosto y vino.
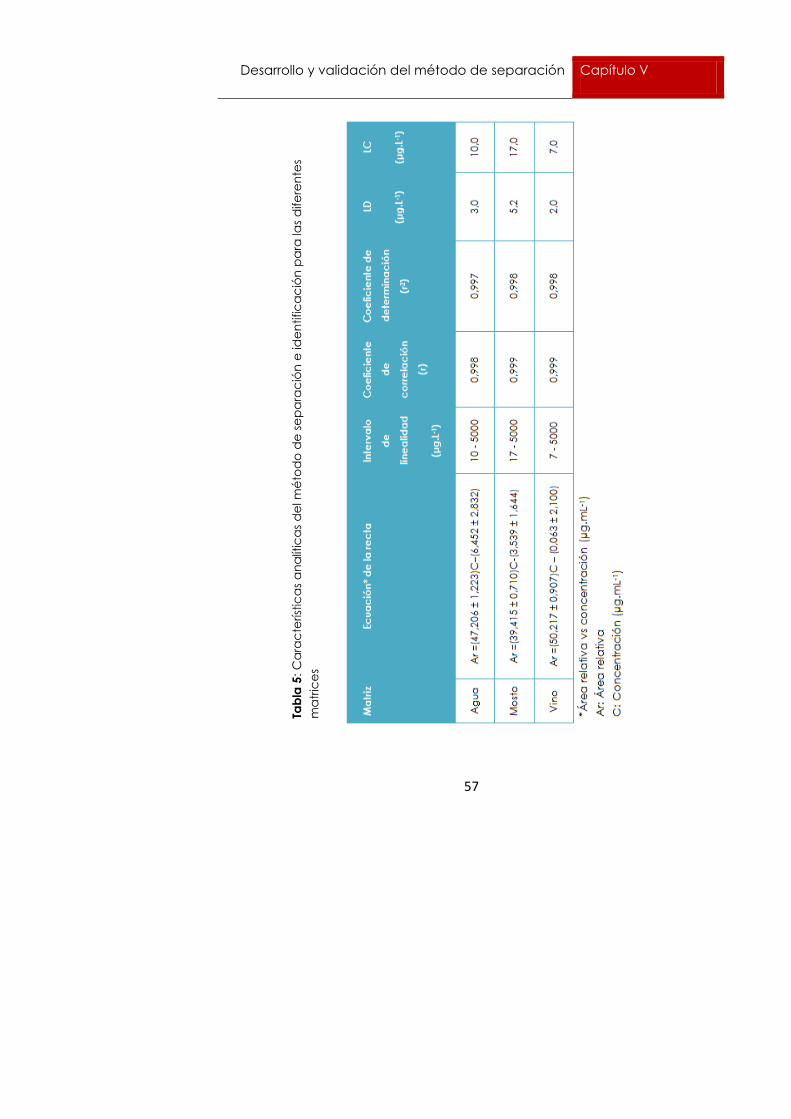
En la tabla 5 se pueden observar las ecuaciones de las curvas de calibrado
para las diferentes matrices objeto de estudio junto con el intervalo de
linealidad y los coeficientes de ajuste lineal.
Desarrollo y validación del método de separación Capítulo V
57
Tab
la 5
: C
ara
cte
ríst
ica
s a
na
lític
as
de
l mé
tod
o d
e s
ep
ara
ció
n e
ide
ntific
ac
ión
pa
ra la
s d
ife
ren
tes
ma
tric
es

Capítulo V Desarrollo y validación del método de separación
58
Como se puede observar, todas las matrices presentaron un coeficiente de
ajuste lineal elevado, con un intervalo de linealidad que incluye el LMR
permitido para este fungicida (5000 µg.L-1). Por tanto se puede concluir que
este método es adecuado para el análisis de dicho fungicida, ya que
permite determinar concentraciones dentro de un rango lineal hasta dicho
valor.
5.5.3. Efecto matriz
El efecto matriz es un aumento o disminución no esperada de la respuesta
de los analitos de interés, que se produce por la coelución de otros
componentes presentes en la matriz.
Para estudiar si existe efecto matriz, se realizó el calibrado en las diferentes
matrices objeto de estudio: disolvente, en este caso agua, mosto y vino.
Después se compararon las pendientes obtenidas en mosto y en vino con
la obtenida en el calibrado en disolvente.
La comparación mediante un estudio estadístico de análisis de la varianza
con un nivel de confianza del 95% indicó que existían diferencias
significativas entre las pendientes de las curvas de calibrado realizadas en
mosto frente a disolvente. Sin embargo entre vino y disolvente no se
encontraron diferencias significativas.
Se comprobó la existencia de efecto matriz en mosto y la no existencia en
vino.

Desarrollo y validación del método de separación Capítulo V
59
5.5.4. Límites de detección (LD) y cuantificación (LC)
El límite de detección se define como la concentración más baja de
analito que puede ser detectada en la muestra. Esto se corresponde con
la concentración de analito que produce una relación señal/ruido ≥ 3.
El límite de cuantificación es la concentración más baja de analito que
puede ser determinada en un nivel aceptable de precisión y exactitud.
Esto se corresponde con la concentración de analito que produce una
relación señal/ruido = 10(Dass 2007).
Los valores de los límites de detección y cuantificación obtenidos para las
tres matrices estudiadas se muestran en la tabla 5.
Los valores más altos de LD y LC se obtuvieron para la matriz mosto con
valores 5,2 y 17,0 µg.L-1 respectivamente, mientras que los valores más bajos
de LD y LC se obtuvieron para la matriz vino con valores 2,0 y 7,0 µg.L-1
respectivamente. Los resultados en disolvente, en este caso agua,
presentaron valores intermedios entre ambas matrices siendo los valores de
LD y LC 3,0 y 10,0 µg.L-1 respectivamente. En todos los casos los valores de
LD y LC fueron inferiores al LMR de este fungicida.
5.5.5. Precisión
Se considera que un método es preciso cuando sucesivos análisis de una
misma muestra producen valores distribuidos alrededor de uno central.
La precisión se mide mediante la repetibilidad y la reproducibilidad del
método. Dado que para estudiar la reproducibilidad se tendría que llevar a
cabo el análisis de la muestra por una persona e incluso un laboratorio
diferente, lo que se realizó fue un estudio de la precisión intermedia.
Capítulo V Desarrollo y validación del método de separación
60
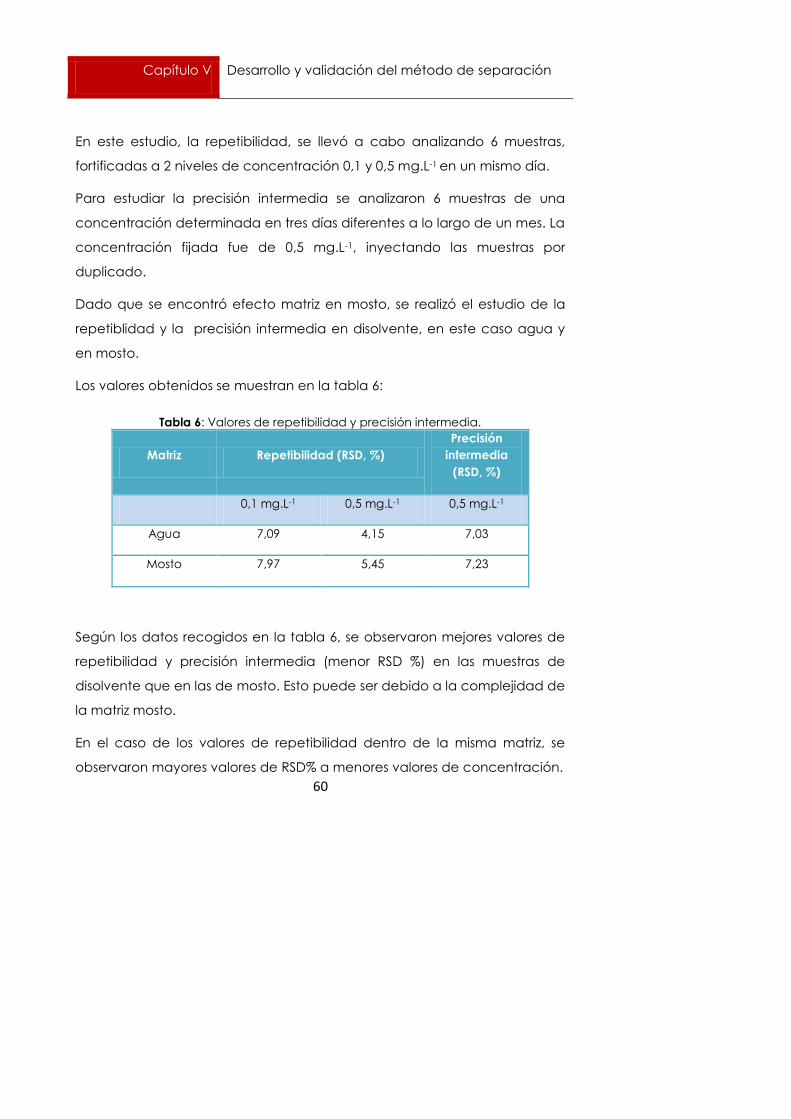
En este estudio, la repetibilidad, se llevó a cabo analizando 6 muestras,
fortificadas a 2 niveles de concentración 0,1 y 0,5 mg.L-1 en un mismo día.
Para estudiar la precisión intermedia se analizaron 6 muestras de una
concentración determinada en tres días diferentes a lo largo de un mes. La
concentración fijada fue de 0,5 mg.L-1, inyectando las muestras por
duplicado.
Dado que se encontró efecto matriz en mosto, se realizó el estudio de la
repetiblidad y la precisión intermedia en disolvente, en este caso agua y
en mosto.
Los valores obtenidos se muestran en la tabla 6:
Tabla 6: Valores de repetibilidad y precisión intermedia.
Matriz Repetibilidad (RSD, %)
Precisión
intermedia
(RSD, %)
0,1 mg.L-1 0,5 mg.L-1 0,5 mg.L-1
Agua 7,09 4,15 7,03
Mosto 7,97 5,45 7,23
Según los datos recogidos en la tabla 6, se observaron mejores valores de
repetibilidad y precisión intermedia (menor RSD %) en las muestras de
disolvente que en las de mosto. Esto puede ser debido a la complejidad de
la matriz mosto.
En el caso de los valores de repetibilidad dentro de la misma matriz, se
observaron mayores valores de RSD% a menores valores de concentración.

Desarrollo y validación del método de separación Capítulo V
61
Al comparar los valores de las muestras de concentración 0,5 mg.L-1
obtenidas el mismo día y en diferentes días se obtuvieron valores mayores
de (RSD %) al realizar los análisis en distintos días. En este caso la variable
tiempo sí que afectó al análisis de las muestras.

CAPÍTULO VI:
FOTODEGRADACIÓN DE
AMETOCTRADÍN


Fotodegradación de Ametoctradín Capítulo VI
65
6. Fotodegradación de Ametoctradín en laboratorio
El objetivo de los estudios de degradación “in vitro” consiste en simular las
condiciones medioambientales a las que se ven expuestos los fungicidas
una vez empleados en el tratamiento del campo. Para ello, se somete al
compuesto seleccionado a los efectos de diferentes agentes de
degradación y se realiza un seguimiento del mismo.
En este caso, Ametoctradrín se vio sometido a fotólisis, producida por la
irradiación de la luz proveniente del simulador solar y a hidrólisis producida
por el tratamiento de la muestra en disolución acuosa a distintos valores de
pH.
La concentración de Ametoctradín empleada en el estudio de las
degradaciones se correspondió con el Límite Máximo de Residuos
permitido en uvas. Se trabajó con una concentración elevada de
fungicida para poder identificar una mayor cantidad de productos de
degradación. Sin embargo al emplear altas concentraciones de fungicida
con muy baja solubilidad en agua, fue necesario el empleo de disolventes
orgánicos para facilitar su disolución. El empleo de estos disolventes
orgánicos puede hacer variar el comportamiento del fungicida respecto al
medio acuoso natural en el que se disuelve para su aplicación en
tratamientos de cultivo en campo.
6.1. Condiciones experimentales
Según trabajos de degradación realizados anteriormente en el grupo de
investigación (Solano Olivan (2009), Plaza Medina (2003)), se establecieron
las condiciones de trabajo del simulador solar trabajando a una potencia
máxima de 80W.

Capítulo VI Fotodegradación de Ametoctradín
66
La cubeta porta muestras con caras de cuarzo se situó a una distancia de
5 cm del simulador solar y una altura óptima para que el haz de luz
atravesase la muestra correctamente, tal y como se indicó en el apartado
4.1.2. del Capítulo IV de esta memoria.
Las fotodegradaciones de Ametoctradín se realizaron en medio
hidroalcohólico, para conseguir una disolución total del compuesto. El
estudio se llevó a cabo variando el pH del medio, estudiando así su
comportamiento a pH ácidos, a pH neutro y a pH básico.
Con el objetivo de evaluar la influencia sobre la degradación del fungicida
de los compuestos propios de la matriz uva, matriz sobre la que se aplica el
tratamiento con Ametoctradín en campo, se realizó la fotodegradación en
mosto real de uvas sin tratar y posteriormente fortificado. El mosto es una
matriz acuosa con elevado contenido en azúcares, polifenoles, proteínas,
aminoácidos, etc.
6.2. Metodología
En matraces de 25 ml se prepararon disoluciones de Ametoctradín en
concentraciones similares al LMR (5 mg.L-1), todas ellas con un contenido
en alcohol metílico del 50% (v/v). Las disoluciones a estudiar fueron:
disolución ácida a pH 2,0 y 3,5, disolución neutra a pH 7,0, disolución
básica a pH 11,0 y mosto.
La muestra a estudiar se colocó en una celda de cuarzo (transparente a la
radiación ultravioleta), manteniéndose en irradiación con la lámpara de
xenón, en ausencia total de luz. Para realizar el seguimiento de la
degradación, se tomaron muestras de 1mL durante distintos periodos de
tiempo, los cuales dependieron del medio en el que se estaba llevando a
cabo la degradación. En las degradaciones de pH ácido y básico se tomó
muestra cada hora durante las primeras 10 horas. En el caso de la

Fotodegradación de Ametoctradín Capítulo VI
67
degradación a pH neutro la toma de muestra se prolongó hasta las 17
horas.
A cada una de estas muestras se les añadió la cantidad correspondiente
de patrón interno, se filtraron a través de un filtro de 0,22 µm y se inyectaron
en el LC-MS para estudiar la evolución de Ametoctradín y la formación de
los principales productos de fotodegradación.
Otro factor tenido en cuenta durante el seguimiento de la
fotodegradación fue el posible aumento de la temperatura de la muestra
debido a la irradiación prolongada de la misma. La comprobación de ese
posible incremento se realizó midiendo la temperatura al inicio y al final de
la degradación no observándose un aumento significativo de la misma.
Durante todo el proceso de degradación la temperatura se mantuvo a
21±3 ºC.
6.3. Cinéticas de fotodegradación de Ametoctradín
En este apartado se recogen los resultados de las cinéticas de
degradación obtenidos en las fotodegradaciones llevadas a cabo en el
laboratorio para Ametoctradín en disoluciones acuosas a distintos valores
de pH y en mosto.
El valor de la constante de velocidad, k, para cada una de las
degradaciones se calculó empleando las ecuaciones de ajuste ya
explicadas en el Capítulo II de esta memoria.
6.3.1. Fotodegradación a pH ácidos
Siguiendo el método de fotodegradación descrito en el apartado anterior,
se llevó a cabo la degradación del fungicida en disolución hidroalcohólica
a pH 2,0 y 3,5.

Capítulo VI Fotodegradación de Ametoctradín
68
Se realizó un seguimiento durante 10 horas en el caso de pH 2,0 y durante
10,5 horas en el caso de pH 3,5. Para esos tiempos, Ametoctradín se había
degradado un 99,71%, degradación prácticamente total a pH=2,0. A pH
3,5 se había degradado un 94,57% para el mismo tiempo.
En las gráficas de las figuras 24 y 25 se representan A) la evolución y B) la
cinética de degradación del Ametoctradín a pH 2,0 y 3,5 respectivamente.
Figura 24: Evolución (A) y cinética (B) de fotodegradación del Ametoctradín
irradiando con lámpara de arco de xenón a 80 W en medio ácido, pH 2,0.
Figura 25: Evolución (A) y cinética (B) de fotodegradación del Ametoctradín
irradiando con lámpara de arco de xenón a 80 W en medio ácido, pH 3,5.
0
20
40
60
80
100
120
0 2 4 6 8 10
% p
ersi
sten
cia
tiempo (h)
A)
0
20
40
60
80
100
120
0 2 4 6 8 10
% p
ers
iste
nci
a
tiempo (h)
A)
0
1
2
3
4
5
6
0 2 4 6 8 10
Ln (
ct)
-Ln
(ci
)
tiempo (h)
B)
-2
-1
0
1
2
3
4
5
6
0 2 4 6 8 10
Ln (
Ct)
- Ln
(C
i)
tiempo (h)
B)

Fotodegradación de Ametoctradín Capítulo VI
69
A partir de los resultados experimentales y teniendo en cuenta las
ecuaciones del Capítulo II, se calculó la constante cinética para cada
proceso.
En la tabla 7 se encuentran los parámetros cinéticos de degradación a pH
ácidos, 2,0 y 3,5.
Tabla 7: Parámetros de las cinéticas de degradación en disolución acuosa a pH 2,0
y 3,5.
pH Ecuación de velocidad t (h) r k (s-1)
x10-5
t1/2
(h)
2,0 Ln (Ct/Ci)=(-0,263±0,040)t+(4,755±0,092)
Ln (Ct/Ci)=(-0,821±0,047)t+(7,073±0,317)
0-4,0
4,0-10,0
0,957
0,993
7,345 ±1,094
22,810±1,309
2,67
0,84
3,5 Ln (Ct/Ci) = (-0,302±0,060)t+(4,721±0,035) 0–10,5 0,998 8,398 ± 0,164 2,31
Se puede observar que la degradación se ajustó a una cinética de primer
orden para ambos valores de pH. En el caso de pH 2,0, se obtuvieron dos
procesos cinéticos según el tiempo de irradiación transcurrido,
presentando una cinética o degradación más acusada a medida que
aumentaba el tiempo de irradiación. En el caso de pH 3,5, se obtuvo una
cinética de primer orden cuya constante presentaba un valor intermedio a
las obtenidas en las cinéticas a pH 2,0.
Para ambos valores de pH se obtuvieron valores bajos de t1/2. Esto indicaría
una degradación acusada a valores de pH ácidos.
Se midieron los valores de pH al dar por finalizado el proceso obteniendo
valores de pH de 2,0 y 2,7 para pH= 2,0 y 3,5 respectivamente.
6.3.2. Fotodegradación a pH neutro
Una vez estudiada la degradación de Ametoctradín a pH ácidos, se
estudió su comportamiento a pH neutro, pH = 7,0.
Capítulo VI Fotodegradación de Ametoctradín
70
El procedimiento fue el mismo que el empleado para las degradaciones
en medio ácido.
Los resultados obtenidos para la degradación en medio neutro aparecen
en las gráficas de la figura 26:
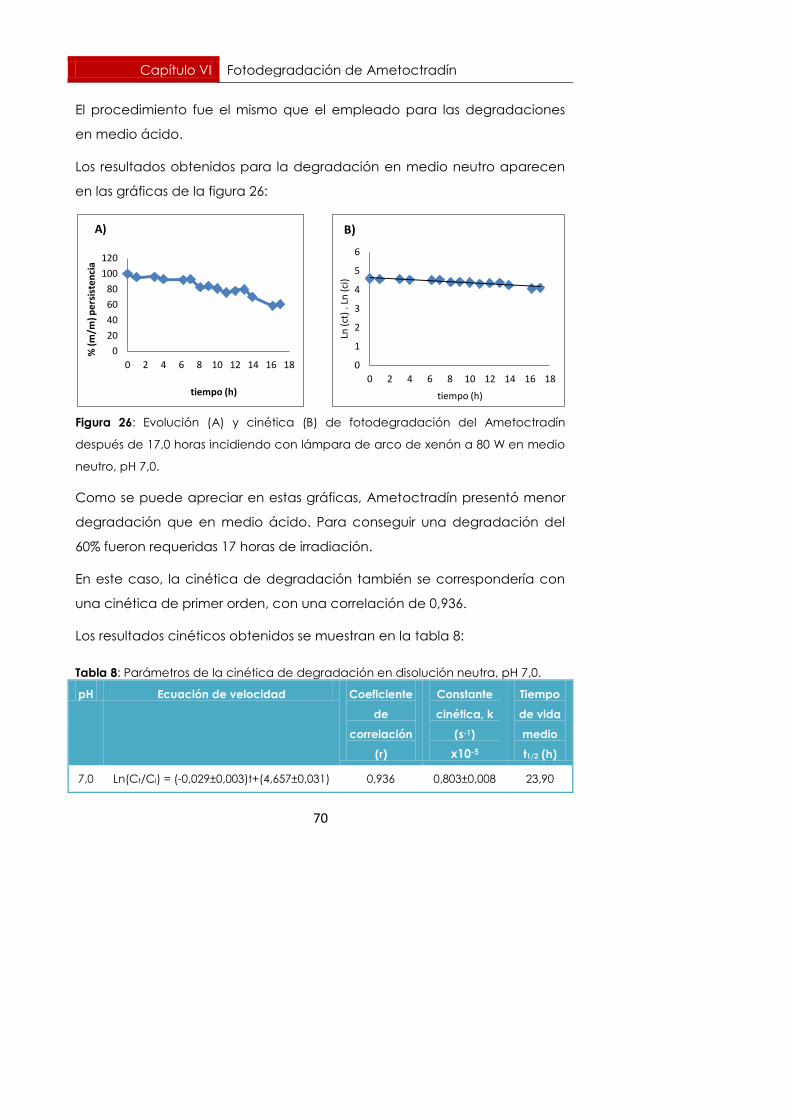
Figura 26: Evolución (A) y cinética (B) de fotodegradación del Ametoctradín
después de 17,0 horas incidiendo con lámpara de arco de xenón a 80 W en medio
neutro, pH 7,0.
Como se puede apreciar en estas gráficas, Ametoctradín presentó menor
degradación que en medio ácido. Para conseguir una degradación del
60% fueron requeridas 17 horas de irradiación.
En este caso, la cinética de degradación también se correspondería con
una cinética de primer orden, con una correlación de 0,936.
Los resultados cinéticos obtenidos se muestran en la tabla 8:
Tabla 8: Parámetros de la cinética de degradación en disolución neutra, pH 7,0.
pH Ecuación de velocidad Coeficiente
de
correlación
(r)
Constante
cinética, k
(s-1)
x10-5
Tiempo
de vida
medio
t1/2 (h)
7,0 Ln(Ct/Ci) = (-0,029±0,003)t+(4,657±0,031) 0,936 0,803±0,008 23,90
0
20
40
60
80
100
120
0 2 4 6 8 10 12 14 16 18
% (
m/m
) pe
rsis
ten
cia
tiempo (h)
A)
0
1
2
3
4
5
6
0 2 4 6 8 10 12 14 16 18
Ln (
ct)
. Ln
(ci
)
tiempo (h)
B)
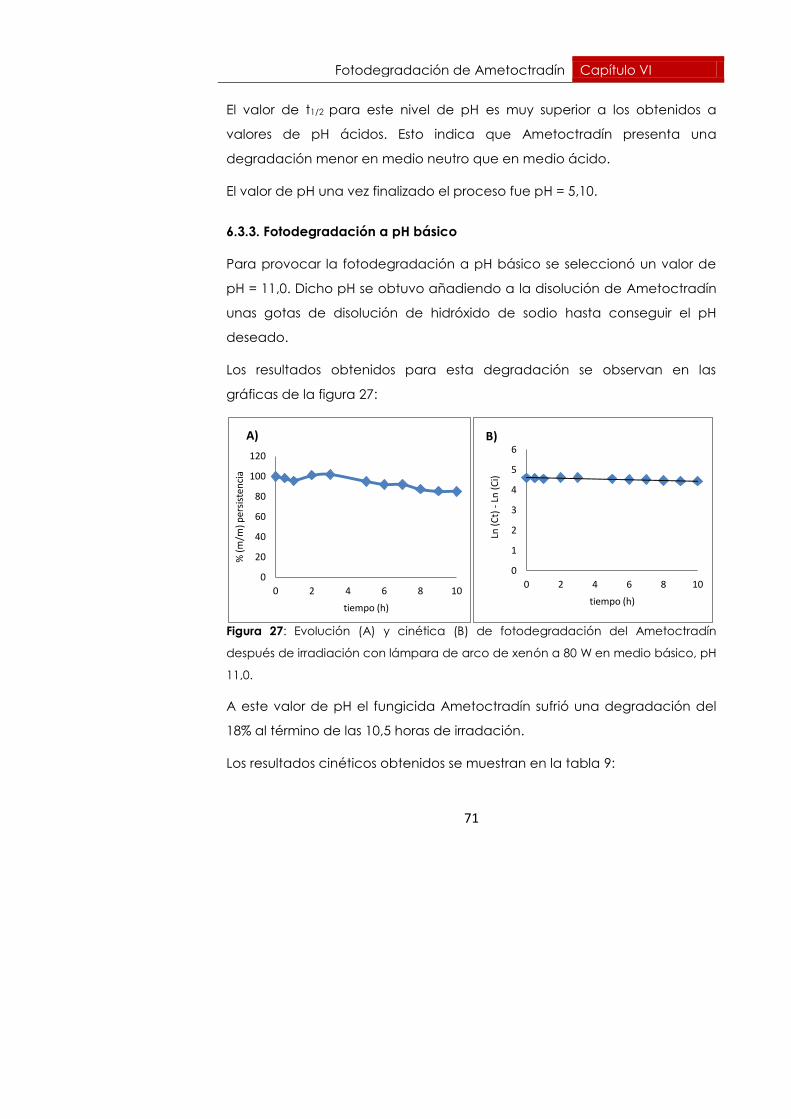
Fotodegradación de Ametoctradín Capítulo VI
71
El valor de t1/2 para este nivel de pH es muy superior a los obtenidos a
valores de pH ácidos. Esto indica que Ametoctradín presenta una
degradación menor en medio neutro que en medio ácido.
El valor de pH una vez finalizado el proceso fue pH = 5,10.
6.3.3. Fotodegradación a pH básico
Para provocar la fotodegradación a pH básico se seleccionó un valor de
pH = 11,0. Dicho pH se obtuvo añadiendo a la disolución de Ametoctradín
unas gotas de disolución de hidróxido de sodio hasta conseguir el pH
deseado.
Los resultados obtenidos para esta degradación se observan en las
gráficas de la figura 27:
Figura 27: Evolución (A) y cinética (B) de fotodegradación del Ametoctradín
después de irradiación con lámpara de arco de xenón a 80 W en medio básico, pH
11,0.
A este valor de pH el fungicida Ametoctradín sufrió una degradación del
18% al término de las 10,5 horas de irradación.
Los resultados cinéticos obtenidos se muestran en la tabla 9:
0
20
40
60
80
100
120
0 2 4 6 8 10
% (
m/m
) per
sist
enci
a
tiempo (h)
A)
0
1
2
3
4
5
6
0 2 4 6 8 10
Ln (
Ct)
- L
n (
Ci)
tiempo (h)
B)

Capítulo VI Fotodegradación de Ametoctradín
72
Tabla 9: Parámetros de las cinéticas de degradación en disolución básica, pH 11,0
pH Ecuación de velocidad Coeficiente
de
correlación (r)
Constante
cinética, k
(s-1) x10-5
Tiempo
de vida
medio
t1/2 (h)
11,0 Ln (Ct/Ci) = (-0,018±0,002)t+(4,623±0,014) 0,930 0,503±0,063 38,51
En la tabla 9 se observa que a este valor de pH Ametoctradín se ajustaba a
una cinetica de primer orden (r = 0,930).
La variación de pH en este caso fue de 0,44 unidades obteniendo un valor
de pH final de 10,85.
El valor de t1/2 a pH 11,0 es superior a los obtenidos a pH neutro y ácido, por
tanto Ametoctradín presentaba menor degradación en medio básico.
6.3.4. Fotodegradación en disoluciones tampón
El seguimiento que se realizó del pH durante el proceso de
fotodegradación indicó una disminución del mismo a medida que
transcurría el tiempo de irradiación en medios ácidos y en medio neutro.
Por este motivo, se realizaron dos degradaciones en medios tamponados a
un valor de pH ácido 3,5 y un pH neutro 7,0.
Se realizó un seguimiento durante 11 horas en el caso del pH ácido, ya que
a ese tiempo la degradación del compuesto fue casi total sin utilizar
disolución tampón.
En el caso del pH neutro la irradiación se mantuvo durante 17 horas.
En las gráficas de las figuras 28 y 29 se representan la evolución y la
cinética de degradación de Ametoctradín en las disoluciones tampón a
pH 3,5 y 7,0.
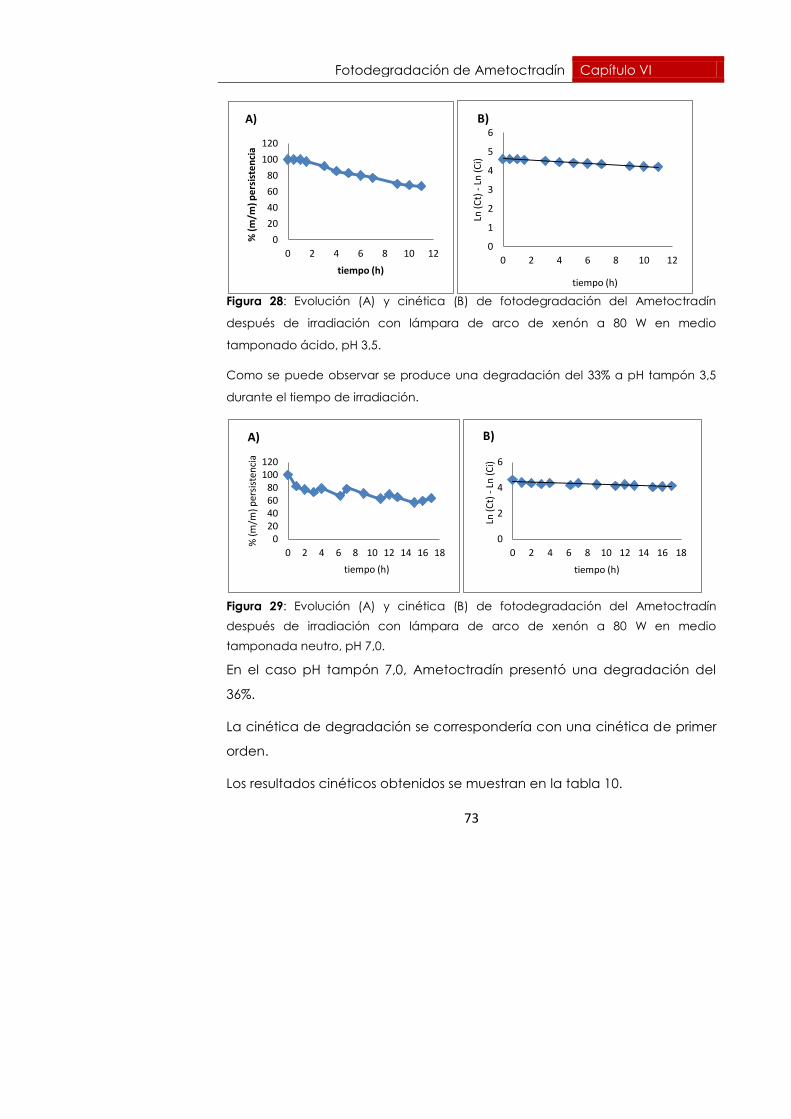
Fotodegradación de Ametoctradín Capítulo VI
73
Figura 28: Evolución (A) y cinética (B) de fotodegradación del Ametoctradín
después de irradiación con lámpara de arco de xenón a 80 W en medio
tamponado ácido, pH 3,5.
Como se puede observar se produce una degradación del 33% a pH tampón 3,5
durante el tiempo de irradiación.
Figura 29: Evolución (A) y cinética (B) de fotodegradación del Ametoctradín
después de irradiación con lámpara de arco de xenón a 80 W en medio
tamponada neutro, pH 7,0.
En el caso pH tampón 7,0, Ametoctradín presentó una degradación del
36%.
La cinética de degradación se correspondería con una cinética de primer
orden.
Los resultados cinéticos obtenidos se muestran en la tabla 10.
0
20
40
60
80
100
120
0 2 4 6 8 10 12 %
(m
/m) p
ers
iste
nci
a
tiempo (h)
A)
0
1
2
3
4
5
6
0 2 4 6 8 10 12
Ln (
Ct)
- L
n (
Ci)
tiempo (h)
B)
0 20 40 60 80
100 120
0 2 4 6 8 10 12 14 16 18
% (
m/m
) per
sist
enci
a
tiempo (h)
A)
0
2
4
6
0 2 4 6 8 10 12 14 16 18
Ln (
Ct)
- L
n (
Ci)
tiempo (h)
B)

Capítulo VI Fotodegradación de Ametoctradín
74
Tabla 10: Parámetros de las cinéticas de degradación en disolución tamponada
ácida, pH 3,5 y neutra, pH 7,0.
pH Ecuación de velocidad Coeficiente
de
correlación
(r)
Constante
cinética, k
(s-1) x10-5
Tiempo
de vida
medio
t1/2 (h)
3,5 Ln (Ct/Ci) = (-0,040±0,001)t+(4,627±0,007) 0,996 1,117±0,033 17,33
7,0 Ln (Ct/Ci) = (-0,022±0,004)t+(4,445±0,035) 0,864 0,605±0,098 31,51
El valor de t1/2 obtenido a pH 3,5 fue menor que el obtenido a pH 7,0,
presentando por tanto mayor degradación a valores más ácidos de pH.
El pH de las disoluciones al final del proceso de irradiación no sufrió
modificación.
6.3.5. Fotodegradación en mosto
En este punto se realizó una degradación en una matriz real mosto,
procedente de uvas tintas. Se observó que Ametoctradín prácticamente
no presentaba degradación en mosto, aproximadamente se degradaba
un 3% después de 11 horas de irradiación.
En la gráfica de la figura 30 se representa la evolución de Ametoctradín en
mosto real.
Figura 30: Evolución de Ametoctradín después de irradiación con lámpara de arco
de xenón a 80 W en mosto real.
0
20
40
60
80
100
120
0 2 4 6 8 10
% (
m/m
) per
sist
enci
a
tiempo (h)
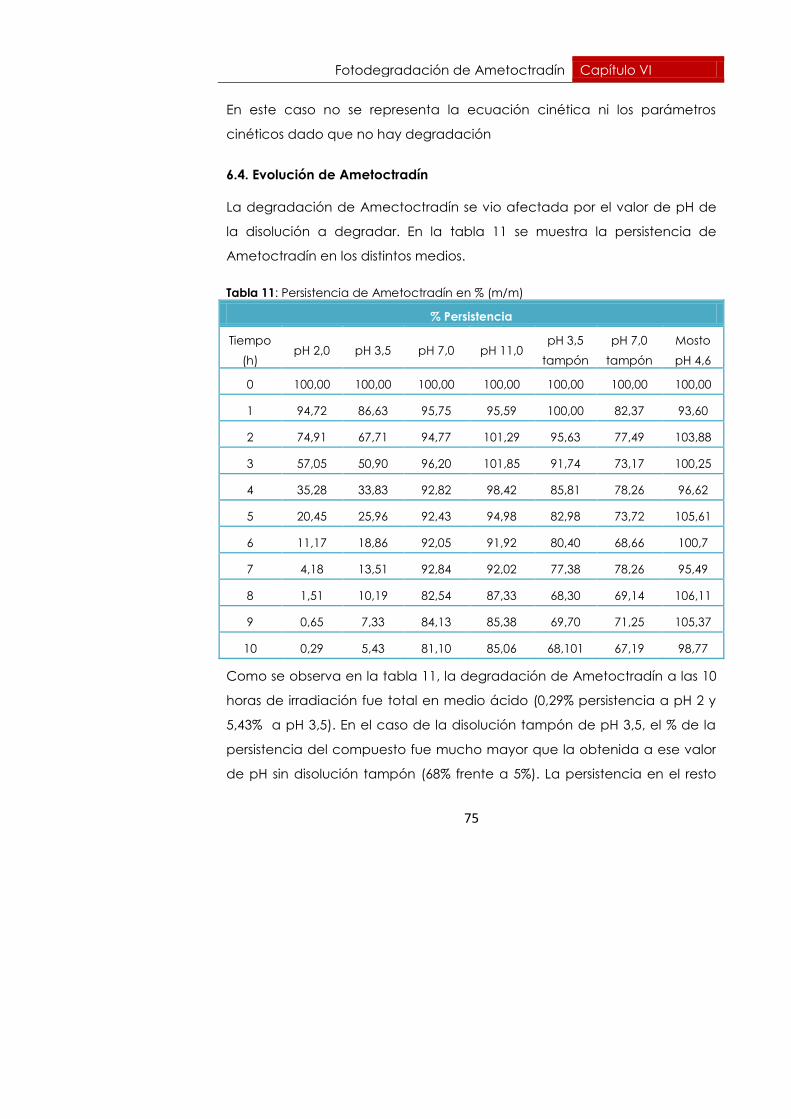
Fotodegradación de Ametoctradín Capítulo VI
75
En este caso no se representa la ecuación cinética ni los parámetros
cinéticos dado que no hay degradación
6.4. Evolución de Ametoctradín
La degradación de Amectoctradín se vio afectada por el valor de pH de
la disolución a degradar. En la tabla 11 se muestra la persistencia de
Ametoctradín en los distintos medios.
Tabla 11: Persistencia de Ametoctradín en % (m/m)
% Persistencia
Tiempo
(h) pH 2,0 pH 3,5 pH 7,0 pH 11,0
pH 3,5
tampón
pH 7,0
tampón
Mosto
pH 4,6
0 100,00 100,00 100,00 100,00 100,00 100,00 100,00
1 94,72 86,63 95,75 95,59 100,00 82,37 93,60
2 74,91 67,71 94,77 101,29 95,63 77,49 103,88
3 57,05 50,90 96,20 101,85 91,74 73,17 100,25
4 35,28 33,83 92,82 98,42 85,81 78,26 96,62
5 20,45 25,96 92,43 94,98 82,98 73,72 105,61
6 11,17 18,86 92,05 91,92 80,40 68,66 100,7
7 4,18 13,51 92,84 92,02 77,38 78,26 95,49
8 1,51 10,19 82,54 87,33 68,30 69,14 106,11
9 0,65 7,33 84,13 85,38 69,70 71,25 105,37
10 0,29 5,43 81,10 85,06 68,101 67,19 98,77
Como se observa en la tabla 11, la degradación de Ametoctradín a las 10
horas de irradiación fue total en medio ácido (0,29% persistencia a pH 2 y
5,43% a pH 3,5). En el caso de la disolución tampón de pH 3,5, el % de la
persistencia del compuesto fue mucho mayor que la obtenida a ese valor
de pH sin disolución tampón (68% frente a 5%). La persistencia en el resto

Capítulo VI Fotodegradación de Ametoctradín
76
de valores de pH fue muy elevada, no observándose degradación en
mosto (98% de persistencia después de 10 horas de irradiación)
6.5. Evolución de los productos de degradación
La determinación de los productos de degradación de fungicidas es una
tarea complicada debido a diferentes factores:
La baja concentración en la que se forman los productos de
degradación.
La dificultad de extraer, separar y analizar los productos de
degradación.
La disposición de las técnicas analíticas adecuadas para estos
compuestos.
En este trabajo, el estudio de los productos de degradación se realizó con
la técnica de cromatografía líquida acoplada a espectrometría de masas
a tiempo de vuelo con ionización por electrospray (ESI-TOF).
Para facilitar la detección de esos productos de degradación, se llevó a
cabo la concentración de las muestras obtenidas al final de cada
degradación. Para ello se empleó un rotavapor equipado con un baño de
agua a 30 ºC. Las muestras se llevaron hasta sequedad y fueron disueltas
en metanol.
Dado que sólo se obtuvo una degradación total de Ametoctradín a pH
ácido, el estudio de los productos de degradación resultantes se llevó a
cabo a pH 2,0 y 3,5.
Para comprobar el posible efecto de la temperatura se realizó la
degradación térmica de una disolución de Ametoctradín a pH 2,0 y a una
temperatura de 50±3ºC durante un tiempo de 10 horas. No se observó

Fotodegradación de Ametoctradín Capítulo VI
77
disminución en la concentración en estas condiciones y durante ese
tiempo.
Una vez comprobada la existencia de los diferentes compuestos se
comprobó su presencia en todas las disoluciones estudias.
6.5.1. pH 2
De forma general, la identificación de los productos de degradación se
realiza en diferentes pasos.
En el primero de ellos se llevó a cabo la comparación del TIC (total ion
chromatogram) de la muestra a tiempo cero de irradiación, con el TIC de
la muestra obtenida después de 10 horas de irradiación. Al realizar dicha
comparación se observó la aparición de picos en la muestra a tiempo final
de irradiación debidos exclusivamente a los nuevos compuestos que
aparecen.
En la figura 31 se muestra la comparación de los TIC de ambas muestras.
Figura 31: Comparación de los TIC obtenidos en las muestras a t=o (color rojo) y a
t=10 horas (color azul) de irradiación a pH 2,0.

Capítulo VI Fotodegradación de Ametoctradín
78
El segundo paso consistió en la comparación de los cromatogramas a
tiempo inicial y final de la degradación extrayendo los picos principales
con la opción “Base Peak Chromatogram”.
La figura 32 muestra la comparación de “Base Peak Chromatogram” de
dichas muestras.
Figura 32: Comparación de los “Base Peak Chromatogram” obtenidos en las
muestras a t=0 (rojo) y a t=10h (azul) de irradiación a pH 2,0.
Las figuras 33 y 34 muestran los picos cromatográficos correspondientes
tanto a disminución de Ametoctradín como a la aparición y aumento de
los productos de degradación genereados durante el periodo de
fotodegradación a los tiempos, t= 0, 3h, 6h y 10h.
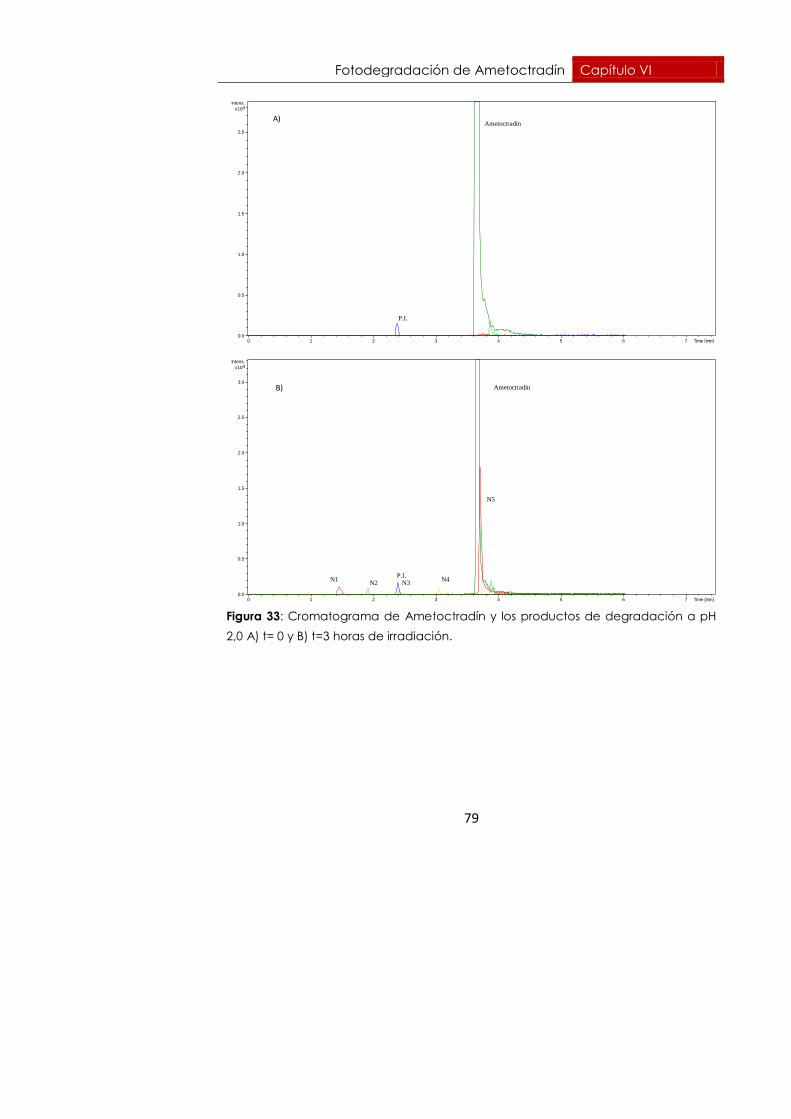
Fotodegradación de Ametoctradín Capítulo VI
79
Figura 33: Cromatograma de Ametoctradín y los productos de degradación a pH
2,0 A) t= 0 y B) t=3 horas de irradiación.
0 1 2 3 4 5 6 7 Time [min]0.0
0.5
1.0
1.5
2.0
2.5
4x10
Intens.
DG PH2.0 FOTO 0.0h_2-b,5_01_12918.d: EIC 215.0900±0.0200 +All MS DG PH2.0 FOTO 0.0h_2-b,5_01_12918.d: EIC 187.0600±0.0200 +All MS
DG PH2.0 FOTO 0.0h_2-b,5_01_12918.d: EIC 276.2200±0.0200 +All MS DG PH2.0 FOTO 0.0h_2-b,5_01_12918.d: EIC 164.0900±0.0200 +All MS
DG PH2.0 FOTO 0.0h_2-b,5_01_12918.d: EIC 250.1700±0.0200 +All MS DG PH2.0 FOTO 0.0h_2-b,5_01_12918.d: EIC 265.1700±0.0200 +All MS
DG PH2.0 FOTO 0.0h_2-b,5_01_12918.d: EIC 294.2300±0.0200 +All MS
0 1 2 3 4 5 6 7 Time [min]0.0
0.5
1.0
1.5
2.0
2.5
3.0
4x10
Intens.
DG PH2.0 FOTO 03.0h_2-a,8_01_12908.d: EIC 215.0900±0.0200 +All MS DG PH2.0 FOTO 03.0h_2-a,8_01_12908.d: EIC 276.2200±0.0200 +All MS
DG PH2.0 FOTO 03.0h_2-a,8_01_12908.d: EIC 265.1700±0.0200 +All MS DG PH2.0 FOTO 03.0h_2-a,8_01_12908.d: EIC 164.0900±0.0200 +All MS
DG PH2.0 FOTO 03.0h_2-a,8_01_12908.d: EIC 187.0600±0.0200 +All MS DG PH2.0 FOTO 03.0h_2-a,8_01_12908.d: EIC 250.1700±0.0200 +All MS
DG PH2.0 FOTO 03.0h_2-a,8_01_12908.d: EIC 294.2300±0.0200 +All MS
P.I.
Ametoctradín
N1 N2 N3
P.I.
Ametoctradín
N4
N5
A)
B)
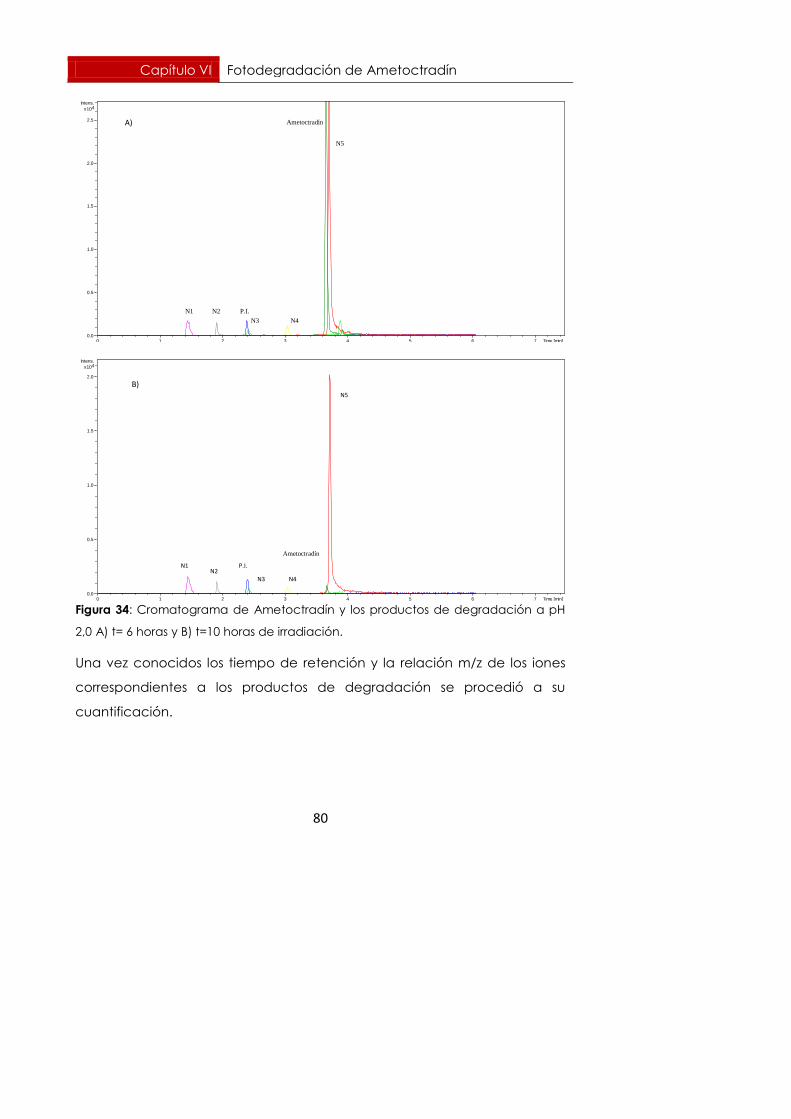
Capítulo VI Fotodegradación de Ametoctradín
80
Figura 34: Cromatograma de Ametoctradín y los productos de degradación a pH
2,0 A) t= 6 horas y B) t=10 horas de irradiación.
Una vez conocidos los tiempo de retención y la relación m/z de los iones
correspondientes a los productos de degradación se procedió a su
cuantificación.
0 1 2 3 4 5 6 7 Time [min]0.0
0.5
1.0
1.5
2.0
2.5
4x10
Intens.
DG PH2.0 FOTO 06.0h_2-a,5_01_12902.d: EIC 215.0900±0.0200 +All MS DG PH2.0 FOTO 06.0h_2-a,5_01_12902.d: EIC 276.2200±0.0200 +All MS
DG PH2.0 FOTO 06.0h_2-a,5_01_12902.d: EIC 164.0900±0.0200 +All MS DG PH2.0 FOTO 06.0h_2-a,5_01_12902.d: EIC 187.0500±0.0200 +All MS
DG PH2.0 FOTO 06.0h_2-a,5_01_12902.d: EIC 250.1700±0.0200 +All MS DG PH2.0 FOTO 06.0h_2-a,5_01_12902.d: EIC 265.1700±0.0200 +All MS
DG PH2.0 FOTO 06.0h_2-a,5_01_12902.d: EIC 294.2300±0.0200 +All MS
0 1 2 3 4 5 6 7 Time [min]0.0
0.5
1.0
1.5
2.0
4x10
Intens.
DG PH2.0 FOTO 10.0h_2-a,1_01_12894.d: EIC 215.0900±0.0200 +All MS DG PH2.0 FOTO 10.0h_2-a,1_01_12894.d: EIC 164.0900±0.0200 +All MS
DG PH2.0 FOTO 10.0h_2-a,1_01_12894.d: EIC 187.0694±0.0200 +All MS DG PH2.0 FOTO 10.0h_2-a,1_01_12894.d: EIC 250.1790±0.0200 +All MS
DG PH2.0 FOTO 10.0h_2-a,1_01_12894.d: EIC 265.1900±0.0200 +All MS DG PH2.0 FOTO 10.0h_2-a,1_01_12894.d: EIC 294.2300±0.0200 +All MS
DG PH2.0 FOTO 10.0h_2-a,1_01_12894.d: EIC 276.2200±0.0200 +All MS
N1 N2 P.I.
N3 N4
Ametoctradín
N5
N1 N2
P.I.
N3 N4
N5
Ametoctradín
A)
B)

Fotodegradación de Ametoctradín Capítulo VI
81
En la figura 35 aparecen las gráficas donde se muestran los metabolitos
encontrados a pH 2,0.
Figura 35: A)Evolución de los productos de degradación y B) Evolución conjunta de
los productos de degradación con Ametoctradín a pH 2,0.
Los primeros metabolitos aparecieron después de una hora de
irradicación. El metabolito mayoritario fue N5 con un porcentaje máximo
de formación de 10% a las 6 horas de irradiación. La tabla 12 recoge los
resultados de los diferentes fotoproductos generados a pH 2,0.

Capítulo VI Fotodegradación de Ametoctradín
82
Tabla 12: Tiempos de retención, porcentajes de formación máximos y m/z
correspondientes a los iones de los principales metabolitos.
Compuesto m/z tr (min) % formación
N 1 164,09 1,44 1,09
N 2 187,06 1,90 0,43
N 3 294,23 2,40 0,32
N 4 250,17 3,04 0,51
Ametoctradín 276,22 3,60 -
N 5 265,17 3,70 10,17
A la vista de los datos recogidos en la tabla anterior se puede concluir que
N5 con una relación m/z de 265,17 fue el metabolito mayoritario para la
fotodegradación de Ametoctradín a pH 2,0.
6.5.2. pH 3,5
Se siguió el mismo procedimiento que para la obtención de los productos
de degradación a pH 2,0.
Las figuras 36 y 37 muestran los picos cromatográficos correspondientes
tanto a disminución de Ametoctradín como a la aparición y aumento de
los productos de degradación genereados durante el periodo de
fotodegradación a distintos tiempos, t= 0, 4h, 7,5h y 10,5h.
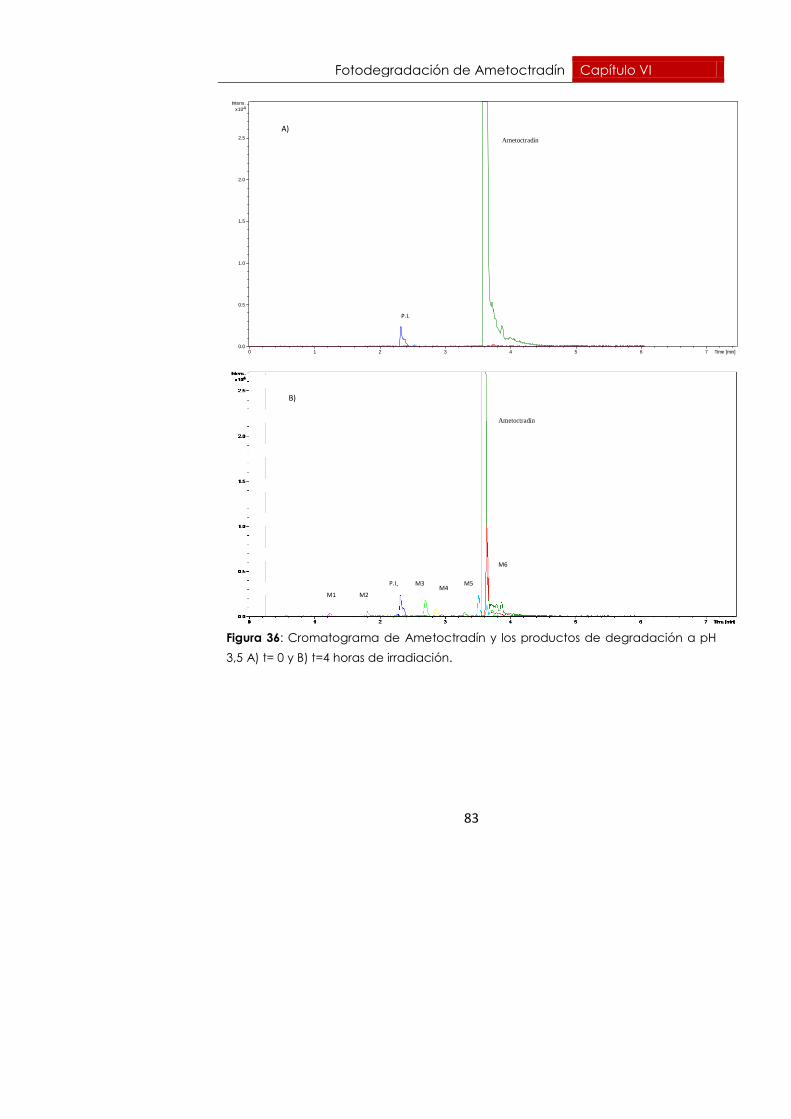
Fotodegradación de Ametoctradín Capítulo VI
83
Figura 36: Cromatograma de Ametoctradín y los productos de degradación a pH
3,5 A) t= 0 y B) t=4 horas de irradiación.
0 1 2 3 4 5 6 7 Time [min]0.0
0.5
1.0
1.5
2.0
2.5
4x10
Intens.
DG PH3.5 0.0 min_2-b,4_01_12090.d: EIC 215.0900±0.0200 +All MS DG PH3.5 0.0 min_2-b,4_01_12090.d: EIC 265.1700±0.0200 +All MS
DG PH3.5 0.0 min_2-b,4_01_12090.d: EIC 266.1700±0.0200 +All MS DG PH3.5 0.0 min_2-b,4_01_12090.d: EIC 308.2400±0.0200 +All MS
DG PH3.5 0.0 min_2-b,4_01_12090.d: EIC 276.2200±0.0200 +All MS DG PH3.5 0.0 min_2-b,4_01_12090.d: EIC 187.0600±0.0200 +All MS
DG PH3.5 0.0 min_2-b,4_01_12090.d: EIC 250.1700±0.0200 +All MS DG PH3.5 0.0 min_2-b,4_01_12090.d: EIC 164.0900±0.0200 +All MS
Ametoctradín
P.I.
M1 M2
P.I. M3 M4
M5
M6
Ametoctradín
A)
B)

Capítulo VI Fotodegradación de Ametoctradín
84
Figura 37: Cromatograma de Ametoctradín y los productos de degradación a pH
3,5 A) t= 7,5 horas y B) t=10,5 horas de irradiación.
0 1 2 3 4 5 6 7 Time [min]0.0
0.5
1.0
1.5
2.0
2.5
4x10
Intens.
DG PH3.5 7.5h_2-a,4_01_12074.d: EIC 215.0900±0.0200 +All MS DG PH3.5 7.5h_2-a,4_01_12074.d: EIC 276.2200±0.0200 +All MS
DG PH3.5 7.5h_2-a,4_01_12074.d: EIC 265.1700±0.0200 +All MS DG PH3.5 7.5h_2-a,4_01_12074.d: EIC 187.0600±0.0200 +All MS
DG PH3.5 7.5h_2-a,4_01_12074.d: EIC 250.1700±0.0200 +All MS DG PH3.5 7.5h_2-a,4_01_12074.d: EIC 266.1700±0.0200 +All MS
DG PH3.5 7.5h_2-a,4_01_12074.d: EIC 308.2400±0.0200 +All MS DG PH3.5 7.5h_2-a,4_01_12074.d: EIC 164.0900±0.0200 +All MS
M1 M2
P.I. M3
M4 M5
M6
Ametoctradín
M1 M2
P.I.
M3
M4
M5
M6
Ametoctradín
A)
B)
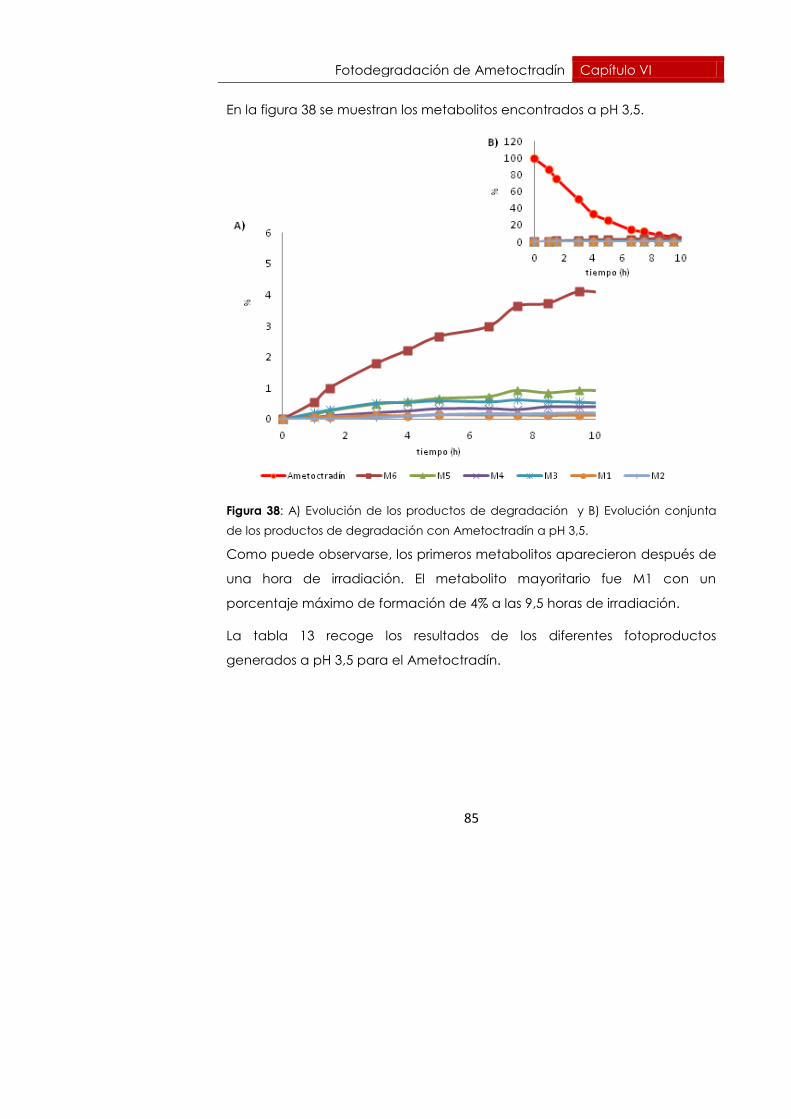
Fotodegradación de Ametoctradín Capítulo VI
85
En la figura 38 se muestran los metabolitos encontrados a pH 3,5.
Figura 38: A) Evolución de los productos de degradación y B) Evolución conjunta
de los productos de degradación con Ametoctradín a pH 3,5.
Como puede observarse, los primeros metabolitos aparecieron después de
una hora de irradiación. El metabolito mayoritario fue M1 con un
porcentaje máximo de formación de 4% a las 9,5 horas de irradiación.
La tabla 13 recoge los resultados de los diferentes fotoproductos
generados a pH 3,5 para el Ametoctradín.

Capítulo VI Fotodegradación de Ametoctradín
86
Tabla 13: Tiempos de retención, porcentajes de formación máximos y m/z
correspondiente a los iones de los principales metabolitos.
Compuesto m/z tr (min) % formación
M 1 164,09 1,25 0,14
M 2 187,06 1,82 0,21
M 3 308,24 2,70 0,63
M 4 250,17 2,86 0,40
M 5 266,17 3,52 0,93
Ametoctradín 276,22 3,60 -
M 6 265,17 3,65 4,10
A la vista de los datos recogidos en la tabla anterior se puede concluir que
M6 con una relación m/z de 265,17 fue el metabolito mayoritario para la
fotodegradación de Ametoctradín a pH 3,5.
Al comparar los metabolitos obtenidos en ambos valores de pH, se observa
que 4 de los compuestos encontrados (m/z 164,09, 187,06, 250,17 y 265,17)
fueron comunes, obteniéndose un mayor porcentaje de los mismos a
valores de pH más ácido.
6.6. Determinación de residuos en el vino
Después de la vendimia, se realizó un tratamiento en los depósitos donde
se iba a llevar a cabo la fermentación alcohólica. Dichos depósitos fueron
tratados con el LMR de Ametoctradín regulado por el Reglamento
396/2005. Cada día durante los 9 días que duró la fermentación alcohólica
se tomaron muestras de vino para estudiar como afectaba la matriz vino
en la evolución del fungicida. Durante ese tiempo, el vino se encontraba
en ausencia total de luz, por lo que no tuvieron lugar procesos de fotólisis.
Para conocer la disipación de Ametoctradín, en este trabajo se analizaron
muestras de las dos variedades de uvas tratadas (graciano y tempranillo)
Fotodegradación de Ametoctradín Capítulo VI
87
tomadas a tiempo cero y a tiempo final de la fermentación alcohólica de
los diferentes depósitos.
Los resultados obtenidos se muestran en la tabla 14:
Tabla 14: Evolución del Ametoctradín durante la fermentación alcohólica.
Variedad Tiempo
(días)
Concentración (mg.L-1)
x 10-2
% (m/m)
Disipación
Graciano 0 8,25±2,09
27,06 ± 6,45
Graciano 9 6,00 ± 0,49
Tempranillo 0 1,28 ± 0,60
20,11 ± 9,56
Tempranillo 9 1,02 ± 0,36
Como se puede observar de estos resultados, la persistencia de
Ametoctradín durante la fermentación fue alta, llegándose a disipar en el
mayor de los casos un 27% (m/m). Hay que tener en cuenta que cuando el
tratamiento se realiza en campo, la disipación se ve favorecida por otros
factores ya comentados (radiación solar, agentes oxidantes, volatilización).
Además de estudiar la evolución de Ametoctradín durante la
fermentación alcohólica se realizó una búsqueda de los productos de
degradación encontrados en el estudio de fotodegradación, no
encontrándose ninguno de ellos en el vino.
6.7 Identificación de los productos de degradación
Una vez conocidas las relaciones masa-carga de los iones
correspondientes a los productos de degradación, se realizó un estudio de
identificación de los mismos.
Mediante una opción del equipo se propuso una fórmula molecular para
cada uno de ellos, indicando su correspondiente error en ppm. El error

Capítulo VI Fotodegradación de Ametoctradín
88
admitido por el equipo es de 5 ppm. Los resultados obtenidos se muestran
en la tabla 15.
Tabla 15: Fórmulas moleculares propuestas para cada ion.
Producto de
degradación
Fórmula
molecular
m/z Error (ppm)
N1 y M1 C7H10N5 164.0931 1.9
N2 y M2 C7H8N4NaO 187.0590 1.2
N3 C15H28N5O 294.2288 3.8
M3 C16H30N5O 308.2445 1.3
N4 y M4 C10H24N3O4 250.1761 8.8
M5 C10H24N3O5 266.1710 4.4
N5 y N6 C10H25N4O4 265.1870 36.2
Según los resultados obtenidos, se pudo identificar la fórmula molecular de
todos ellos excepto los de m/z 250,17 y 265,17 ya que el error para la
fórmula propuesta era mayor que el admitido por el equipo.
Para obtener más información acerca de los productos de degradación se
provocó la ruptura de los iones encontrados mediante la técnica masas-
masas (MS-MS).
Para ello se seleccionó a través del cuadrupolo los iones precursores
deseados y a continuación se les aplicó un voltaje (20-35 V) provocando
su fragmentación.
El espectro MS-MS que muestra la fragmentación del ión 294,23 se recoge
en la figura 39.

Fotodegradación de Ametoctradín Capítulo VI
89
Figura 39: Espectro MS-MS del ion 294,23.
La estructura propuesta para el metabolito con esta relación m/z se
muestra en la figura 40:
m/z 294,23
Figura 40: Estructura propuesta para el metabolito con m/z 294,23
Dicha estructura se correspondía con la fórmula molecular propuesta por
el equipo, siendo el error de 3,8 ppm.
Al observar los fragmentos obtenidos, el ion con m/z de 277,20 coincidiría
con la pérdida del hidróxido. Todos estos resultados deberían confirmarse
por RMN.
179.0996193.1663
210.1922
223.1995
277.2108
293.2205
17.0267
+MS2(294.0000), 20-20eV, 2.36min #280
0
500
1000
1500
2000
2500
3000
Intens.
160 180 200 220 240 260 280 300 320 m/z
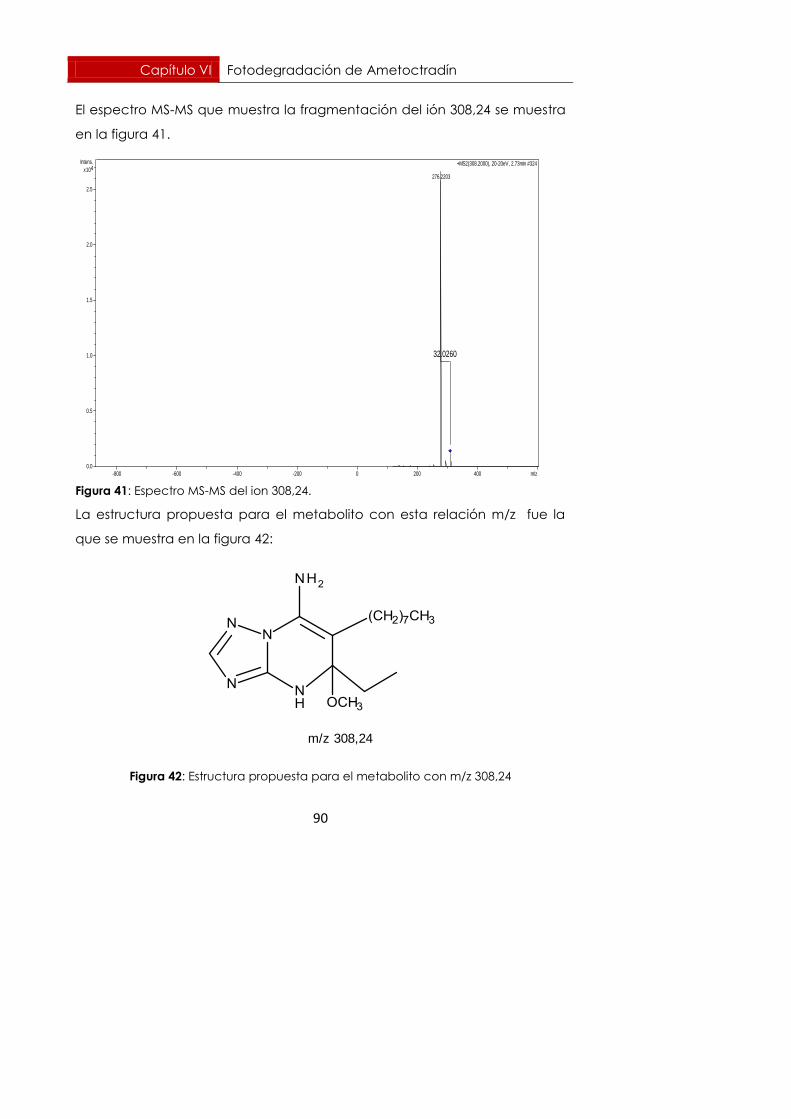
Capítulo VI Fotodegradación de Ametoctradín
90
El espectro MS-MS que muestra la fragmentación del ión 308,24 se muestra
en la figura 41.
Figura 41: Espectro MS-MS del ion 308,24.
La estructura propuesta para el metabolito con esta relación m/z fue la
que se muestra en la figura 42:
m/z 308,24
Figura 42: Estructura propuesta para el metabolito con m/z 308,24
276.2203
32.0260
+MS2(308.2000), 20-20eV, 2.73min #324
0.0
0.5
1.0
1.5
2.0
2.5
4x10
Intens.
-800 -600 -400 -200 0 200 400 m/z

Fotodegradación de Ametoctradín Capítulo VI
91
Dicha estructura se correspondía con la fórmula molecular propuesta por
el equipo, siendo el error de 1,3 ppm.
Al observar los fragmentos obtenidos, el ion con m/z de 276,22 coincidiría
con la pérdida del metóxido y un hidrógeno, obteniéndose la estructura
del compuesto inicial correspondiente a la materia activa.
Con la información obtenida en el estudio de identificación de dos de los
productos de degradación se propuso una parte en la ruta metabólica
para Ametoctradín en disolución hidroalcóholica 50% (v/v) bajo radiación,
la cual se muestra en la figura 43.
m/z 294,23m/z 308,24
m/z 277,22
hv
m/z 276,22
Figura 43: Parte propuesta en la ruta de degradación para Ametoctradín.
Los pasos de esta ruta metabólica consistieron en la protononación inicial
de Ametoctradín debida al medio ácido, seguida de la entrada del grupo
hidroxi o metoxi presentes en el disolvente empleado.

CAPÍTULO VII:
CONCLUSIONES


Conclusiones Capítulo VII
95
Conclusiones
Las conclusiones finales que se han obtenido una vez finalizado el trabajo
experimental y analizado los resultados son las siguientes:
UPLC-MS es una técnica de separación e identificación adecuada
para el análisis de residuos del fungicida Ametoctradín.
El método empleado es selectivo, ya que no aparecen señales
cromatográficas que interfieran en las señales de los analitos.
Además se trata de un método preciso con buenos valores de
repetibilidad y precisión intermedia.
El método empleado permite detectar y cuantificar valores de
Ametoctradín iguales al LMR, siendo los valores de LD y LC inferiores
al LMR.
Se observa la aparición de efecto matriz en mosto, debido a la
existencia de diferencias significativas entre las pendientes
obtenidas de la calibración en mosto y disolvente, en este caso
agua.
En el desarrollo de este trabajo no ha sido necesario ningún
tratamiento previo de la muestra, pudiéndose realizar por inyección
directa.
Ametoctradín presenta cinéticas de fotodegradación de primer
orden a todos los valores de pH estudiados, obteniéndose altos
coeficientes de correlación.
Ametoctradín no presenta fotodegradación en mosto, siendo su
persistencia del 97% después de 11 horas de irradiación.
El mayor porcentaje de degradación se produce a valores ácidos
de pH, obteniéndose una degradación total a las 10 horas de
irradiación.

Capítulo VII Conclusiones
96
Mediante UPLC-MS se han podido detectar y cuantificar los
productos de degradación, siendo el mayoritario el de m/z 265,17,
el cual aparece tanto a pH 2,0 y 3,5 con unos porcentajes máximos
de 10,17% y 4,10% respectivamente.
En el estudio de Ametoctradín en bodega se ha observado que
alcanza un valor máximo de disipación de 27% en los dos tipos de
variedades de uva estudiados: graciano y tempranillo.
Mediante el empleo de MS-MS se han podido identificar dos
productos de degradación gracias a la información obtenida de la
fragmentación de los iones precursores.

CAPÍTULO VIII:
BIBLIOGRAFÍA


Bibliografía Capítulo VIII
99
BIBLIOGRAFÍA
1. Agüera, A., Almansa, E., Tejedor, A., Fernández-Alba, A.R., Malato,
S., Maldonado, M.I. “Photocatalytic pilot scale degradation study of
pyrimethanil and of its main degradation products in waters by
means of solid-phase extraction followed by gas and liquid
chromatography with mass spectrometry detection” (2009)
Environmental Science and Technology 34 (8), pp. 1563-1571.
2. Angioni, A., Garau, A., Caboni, , P., Russo, M.T, Farris, G.A., Zara, S.,
Cabras, P. ”Gas chromatographic ion trap mass spectrometry
determination of zoxamide residues in grape, grape processing and
in the fermentation process” Journal of Chromatography A Volume
1097, Issues 1-2, 2005 ,pages 165-170)
3. Burrows, H.D., Canle L, M., Santaballa, J.A, Steenken, S. “Reaction
pathways and mechanisms of Photodegradation of pesticides”.
Journal of Photochemistry and Photobiology B: Biology 67 (2002) 71-
108.
4. Cabras, P, Angioni, A. “Pesticide Residues in Grapes, Wine and Their
Procesing Products” Journal of Agricultural and Food Chemistry 48
(4), 967-972.
5. Calza, P., Medana, C., Baiocchi, C., Branca, P., Pelizzetti, E.
“Characterisation by high-performance liquid chromatography-
multiple mass spectrometry of intermediate compounds formed
from mepanipyrim photoinduced degradation” (2004)Journal of
Chromatography A 1049 (1-2), pp.115-125.
6. Chen, T., Fu, F., Chen, Z., Li, D., Zhang, L., Chen, G. “Study on the
photodegradation and microbiological degradation of pirimicarb
insecticide by using liquid chromatography coupled with ion-trap

Capítulo VIII Bibliografía
100
mass spectrometry” (2009) Journal of Chromatography A 1216 (15),
pp. 3217-3222.
7. Dass, C. “Fundamentals of contemporary mass spectrometry” Wiley
2007
8. Economou,A., Botitsi, H., Antoniou, S., Tsipi, D. “Determination of
multi-class pesticides in wines by solid-phase extraction and liquid
chromatography-tandem mass spectrometry” Journal of
Chromatography A, 1216 (2009) 5856-5867.
9. Flamini, Ricardo, Panighel, Annanita “Mass Spectrometry in grape
and wine chemistry. Part II. The consumer protection” Wiley
InterScience 2006
10. Fontana, A.R., Rodriguez, I., Ramil, M., Altamirano, J.C., Cela, R.
“LIquid Chromatography time of flight mass spectrometry fallowing
soptive microextraction for the determination of fungicide residues
in wine” Anal Bioanal Chem 2011 401767-775)
11. Gavrilescu, M. “Fate of Pesticides in the environment and its
Bioremediation” Engineering in Life Sciences Volume 5, Issue 6,
pages 497526 (2005).
12. Glosary of terms used in photochemistry 3º Edition (IUPAC
Recommendations 2006)Pure Appl. Chem., Vol. 79, No.3,pp. 293-
465(2007)
13. Hogendoorn, Elbert, Van Zoonen, Piet. “Recent and future
developments of liquid chromatography in pesticide trace analysis”
Review Journal of Chromatography A, 892 (2000) 435–453.
14. Jiménez, J.J, Bernal, J.L., del Nozal, M.J. Arias, E.,Bernal, J., Toribio, L.
“Persistance and degradation of metalaxyl, lindane, fenvalerate
and deltamethrin during the wine making process” Food Chemistry,
104 (2007) 216-223.

Bibliografía Capítulo VIII
101
15. Kot-Wasik, A., Dabrowska, D., Namiesnik, J. ”Stability Studies of
Selected Phenoxyacid Herbicides in Water Samples and
Determination of Their Transformation Products” 2006 Bulletin of
environmental contamination and toxicology 77 (2) pp. 245-251.
16. Kot-Wasik, A., Dabrowska, D., Namiesnik, J.”The importance of
degradation in the fate of selected organic compounds in the
environment. Part I. General Considerations” Polish Journal of
Environmental Studies Vol.13,No.6 (2004) 607-616.
17. Krieguer,M.S, Yoder, R.N,Gibson, R. “Photolytic degradation of
florasulam on soil and in wáter” J.Agric, Food Chem 2000, 48, 3710-
3717.
18. Lagunas-Allué, L., Sanz-Asensio, J., Martínez-Soria, M.T. “Validation of
a microwave-assisted extraction gas chromatography detection
method for the determination of fungicides in grapes” Anal.
Methods, 2011, 3, 2881.
19. Lagunas-Allué, L., Sanz-Asensio, J., Martínez-Soria, M.T., Salvador, A.,
Ferronato, C., Choveron, J.M. “Degradation intermediates and
reaction pathway of pyraclostrobin with TiO2 photocatalysis”
Applied Catalysis B: Environmental 115-116 (2012)285-293.
20. Likas, D.T., Tsiropoulos, N.G., Miliadis, G.E. “Rapid gas
chromatographic method for the determination of famoxadone,
trifloxystrobin and fenhexamid residues in tomato, grape and wine
samples” Journal of Chromatography A, 1150 (2007) 208-214.
21. Merkel, M., Frechen,T., Gold, R.E.,Schiffer, M., Levy, T., Saramago, J.
“Initium: A New Innovative Fungicide of a New Chemical Class for
the Control of Late Blight and Downy Mildew Diseases” Acta
horticulturae (2012)
22. Millán, S., Sampedro, M.C., Unceta, N., Goicolea, M.A., Rodriguez, E.,
Barrio, R.J. “Coupling solid-phase microextraction and high-

Capítulo VIII Bibliografía
102
performance liquid chromatography for direct and sensitive
determination of halogenated fungicides in wine” Journal of
Chromatography A Volume 995, Issues 1-2, 2003, pages 135-142.)
23. Morrica, P., Fidente, P., Seccia, S. “High-performance liquid
chromatographic mass spectrometric identification of the
photoproducts of cymoxanil” (2005) Biomedical
Chromatography 19 (7), pp. 506-512.
24. Plaza Medina, María. “Aislamiento y caracterización de residuos y
matabolitos de pesticidas. Estudios degradativos en muestras
agroalimentarias” (2003).
25. Sandra, P., Tienpont, B., Vercammen, J., Tredoux, A., Sandra,T.,
David,F. ”Stir bar sorptive extraction applied to the determination of
dicarboximide fungicides in wine” Journal of Chromatography A,
928 (2001) 117-126)
26. Solano Oliván, Vanesa “Estudio analítico de fungicidas en matrices
enológicas. Fotodegradación “in vitro” de estrobilurinas aplicadas
en vid” (2009).
27. Vaquero-Fernández, L., Sáenz- Hernández, A., Sanz-Asensio, J.,
Fernández-Zurbano, P., Sainz-Ramirez,M., Pons-Jubera, B., López-
Alonso, M., Martinez-Soria, M.T: “Determination of cyprodinil and
fludioxonil in the fermentative process of must by high-performance
liquid chromatography-diode array detection” Journal of the
Science of food and Agriculture Volume 88, Issue 11, 2008a, pages
1943-1948
28. Vaquero-Fernández, L., Sanz-Asensio, J., Férnandez-Zurbano, P.,
Sainz-Ramírez, M., López-Alonso, M., Epifacio-Fernández, S.I.,
Martínez-Soria, M.T. “Development of a liquid-liquid extraction
method for the determination of pyrimethanil, metalaxyl,

Bibliografía Capítulo VIII
103
dichlofluanid and penconazol during the fermentative process of
must by GC-NPD” Published online in Wiley Interscience 2008b.
29. Vaquero-Fernández, L., Sanz-Asensio,J., López-Alonso, S.I., Martínez-
Soria, M.T. “Fate and distribution of pyrimethanil, metalaxyl,
dichlofluanid and penconazol fungicides from treated grapes
intended for winemaking” Food Additives and Contaminants Vol.26,
No.2, February 2009b, 164-171.
30. Vaquero-Fernández, L., Sanz-Asensio,J., López-Alonso, S.I., Martínez-
Soria, M.T. “Analysis of Pyrimethanil, Metalaxyl, Dichlofluanid, and
Penconazol in Must and Wine from Red Grapes by Solid-Phase
Extraction and Gas Chromatography” Analytical Letters, 42: 1761-
1783, 2009a.
31. Viñas,P., Aguinaga,N., Campillo, N., Hernández-Córdoba, M.
“Comparison of stir bar sorptive extraction and membrane-assisted
solvent extraction for the ultra-performance liquid chromatographic
determination of oxazole fungicide residues in wines and juices”
Journal of chromatography A, 1194 (2008) 178-183.


















