
Alberto Rábano Gutiérrez Nuevos criterios en el...
26
VIII Congreso Virtual Hispanoamericano de Anatomía Patológica — Octubre de 2006 http://conganat.cs.urjc.es Conferencias Invitadas Alberto Rábano Gutiérrez Carmen Guerrero Márquez Unidad de Neuropatología Fundación Hospital Alcorcón Madrid, España. Correspondencia: Dr. Alberto Rábano Gutiérrez Fundación Hospital Alcorcón Avda. de Budapest, 1 28922 Alcorcón Madrid Telf: +34 91 621 98 59 Fax: +34 91 621 94 00 E-mail: [email protected] Nuevos criterios en el diagnóstico histológico de las Demencias El panorama neuropatológico de las demencias ha cambiado radicalmente en los últimos años. La incorporación de nuevos anticuerpos para inmu- notinción y nuevas técnicas moleculares al diagnóstico neuropatológico ha permitido establecer los límites y la heterogeneidad interna de entidades co- mo la Demencia con cuerpos de Lewy y las demencias frontotemporales. La definición de criterios diagnósticos a partir de series amplias de cere- bros ha permitido abordar el problema de la patología combinada y mixta, específicamente en relación con la E. de Alzheimer. En este articulo se re- visan de forma esquemática los actuales criterios neuropatológicos para el diagnóstico de las demencias. Palabras clave: demencias; diagnóstico; neuropatología; criterios The neuropathological knowledge of dementias has suffered a radical change along the last years. New antibodies for immunohistochemistry and new molecular techniques have allowed us to establish the limits and the heterogeneity of entitities like Dementia with Lewy bodies or the Fron- totemporal dementias. The definition of diagnostic criteria based on large series of brains is an invaluable aid in order to approach difficultproblems like combined and mixed pathology, particularly as referred to Alzheimer’s disease. This paper reviews in a schematic way current neuropathological criteria for the diagnosis of dementias. Keywords: dementia; diagnosis; neuropathology; criteria INTRODUCCIÓN Si no esperas lo inesperado, no lo encontrarás... Heráclito, fragmento 18. En este artículo pretendemos dar una visión actualizada del diagnóstico histológico de las demencias, tal como se plantea en el contexto de un banco de cerebros. Explica- remos brevemente los dos sumandos de este binomio. Los neuropatólogos estamos acostumbrados a cargar con el peso del "diagnóstico definitivo" de los casos. Espe- cialmente en las sesiones clínicopatológicas, nos senti- mos a menudo como los técnicos que analizan la caja negra después de una catástrofe aérea. Sin embargo, co- mo se verá a continuación, la evolución de los propios criterios diagnósticos y las técnicas moleculares están convirtiendo el diagnóstico histológico en un momen- to más en el diagnóstico de las demencias, definitivo o cuasi-definitivo en algunos casos, pero parcial o proba- bilista en muchos otros. Como demuestran precisamente las sesiones clínicopatológicas, la clasificación definiti- va (ahora sí) de un caso requiere la integración de toda la información clínica, neurorradiológica, neuropatológica y molecular, si está disponible. La labor de un banco de cerebros va más allá del diag- nóstico neuropatológico. Ya en el momento de la extrac- ción el procedimiento está dirigido a conservar tejido a largo plazo para investigación. Cuando se remite mate- rial a un investigador, el tejido no sólo debe estar diag- nosticado, sino además correctamente clasificado. El es- tadio evolutivo de la enfermedad, la patología combina- da, y rasgos histológicos no relevantes para el diagnósti- co neuropatológico, pero sí para la descripción completa del caso, pueden afectar a los resultados de investigación tanto como la mera clasificación diagnóstica. —1—
Transcript of Alberto Rábano Gutiérrez Nuevos criterios en el...

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Alberto Rábano GutiérrezCarmen Guerrero Márquez
Unidad de NeuropatologíaFundación Hospital AlcorcónMadrid, España.
Correspondencia:Dr. Alberto Rábano GutiérrezFundación Hospital AlcorcónAvda. de Budapest, 128922 Alcorcón Madrid
Telf: +34 91 621 98 59Fax: +34 91 621 94 00E-mail: [email protected]
Nuevos criterios en el diagnóstico histológico de lasDemencias
El panorama neuropatológico de las demencias ha cambiado radicalmenteen los últimos años. La incorporación de nuevos anticuerpospara inmu-notinción y nuevas técnicas moleculares al diagnóstico neuropatológico hapermitido establecer los límites y la heterogeneidad interna de entidades co-mo la Demencia con cuerpos de Lewy y las demencias frontotemporales.La definición de criterios diagnósticos a partir de series amplias de cere-bros ha permitido abordar el problema de la patología combinada y mixta,específicamente en relación con la E. de Alzheimer. En este articulo se re-visan de forma esquemática los actuales criterios neuropatológicos para eldiagnóstico de las demencias.
Palabras clave:demencias; diagnóstico; neuropatología; criterios
The neuropathological knowledge of dementias has suffereda radicalchange along the last years. New antibodies for immunohistochemistryand new molecular techniques have allowed us to establish the limits andthe heterogeneity of entitities like Dementia with Lewy bodies or the Fron-totemporal dementias. The definition of diagnostic criteria based on largeseries of brains is an invaluable aid in order to approach difficultproblemslike combined and mixed pathology, particularly as referred to Alzheimer’sdisease. This paper reviews in a schematic way current neuropathologicalcriteria for the diagnosis of dementias.
Keywords: dementia; diagnosis; neuropathology; criteria
INTRODUCCIÓN
Si no esperas lo inesperado, no lo encontrarás...Heráclito, fragmento 18.
En este artículo pretendemos dar una visión actualizadadel diagnóstico histológico de las demencias, tal como seplantea en el contexto de un banco de cerebros. Explica-remos brevemente los dos sumandos de este binomio.
Los neuropatólogos estamos acostumbrados a cargar conel peso del "diagnóstico definitivo" de los casos. Espe-cialmente en las sesiones clínicopatológicas, nos senti-mos a menudo como los técnicos que analizan la cajanegra después de una catástrofe aérea. Sin embargo, co-mo se verá a continuación, la evolución de los propioscriterios diagnósticos y las técnicas moleculares estánconvirtiendo el diagnóstico histológico en un momen-
to más en el diagnóstico de las demencias, definitivo ocuasi-definitivo en algunos casos, pero parcial o proba-bilista en muchos otros. Como demuestran precisamentelas sesiones clínicopatológicas, la clasificación definiti-va (ahora sí) de un caso requiere la integración de toda lainformación clínica, neurorradiológica, neuropatológicay molecular, si está disponible.
La labor de un banco de cerebros va más allá del diag-nóstico neuropatológico. Ya en el momento de la extrac-ción el procedimiento está dirigido a conservar tejido alargo plazo para investigación. Cuando se remite mate-rial a un investigador, el tejido no sólo debe estar diag-nosticado, sino además correctamente clasificado. El es-tadio evolutivo de la enfermedad, la patología combina-da, y rasgos histológicos no relevantes para el diagnósti-co neuropatológico, pero sí para la descripción completadel caso, pueden afectar a los resultados de investigacióntanto como la mera clasificación diagnóstica.
— 1 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
El panorama de las demencias, tal como puede contem-plarlo un neuropatólogo, no ha dejado de cambiar desdehace algunos años. Los criterios diagnósticos se han he-cho prácticos y operativos, disponemos de técnicas quenos permiten reconocer los principales procesos patoló-gicos en sus distintas fases evolutivas, y ver cómo secombinan creando una diversidad fenotípica cada vezmenos caótica. Grandes grupos de patología, como lasdemencias asociadas a alfa-sinucleinopatía, las demen-cias frontotemporales o las enfermedades priónicas, hansufrido verdaderas revoluciones en su clasificación diag-nóstica.
El presente artículo pretende recoger los principales cri-terios histológicos que utilizamos hic et nunc en el diag-nóstico de las demencias, y se ha estructurado más conintención de revisión actualizada que como un manualpráctico de diagnóstico. No obstante, puesto que de al-guna forma reproduce nuestra estrategia de abordaje decada caso que estudiamos, es posible que tenga algunaaplicabilidad práctica.
Esta estrategia está reflejada en el índice, que incluye,junto a entidades clínico-patológicas establecidas, gru-pos amplios definidos por la proteína patológica asocia-da, tal como la podemos detectar mediante inmunohis-toquímica o con técnicas moleculares como el Westernblot.
PROCEDIMIENTOS BÁSICOS DE UN BANCODE CEREBROS
Un primer proceso clave en el funcionamiento de unbanco de cerebros es la logística de la extracción deltejido mediante autopsia neuropatológica. Mediante unsistema adecuado de guardias y alertas, el banco debegarantizar la extracción en las mejores condiciones parala conservación del tejido y con la mínima incomodidadpara los familiares del paciente. Como en cualquier au-topsia neuropatológica, y aunque los fallecidos sean do-nantes del banco, se deben cumplir los requisitos ético-legales de la autopsia (solicitud y autorización firmada),y es imprescindible hacer una evaluación de bioseguri-dad antes de iniciar la autopsia (tuberculosis, virus dehepatitis, enfermedades priónicas) (1).
La extracción del cerebro se realiza de modo convencio-nal cuando no se sospecha enfermedad priónica. En elcaso de que no pueda descartarse razonablemente estapatología, o de que exista una sospecha clínica efecti-
va, adoptamos medidas de bioseguridad suplementariasa las básicas recomendadas: protección especial de lasinstalaciones y del personal, apertura del cráneo median-te sierra manual y manipulación del tejido fresco y fijadoen cabina de bioseguridad (2). Es conveniente obtener entodos los casos una muestra de líquido cefalorraquídeopara medir el pH, como indicador del estado del tejidotras el periodo agónico y postmórtem. Cuando existendatos de patología de motoneurona u otras patologíasmedulares, se extrae la médula espinal en su totalidad.En nuestra experiencia, el abordaje posterior es preferi-ble cuando la autopsia es estrictamente neuropatológica.En función de las necesidades diagnósticas del caso y delas solicitudes de material recibidas por el banco, pue-den obtenerse muestras adicionales como músculo, ner-vio periférico y ojo, o muestras en fijadores especiales.
Una vez extraído el cerebro, se pesa y se hace el examenmacroscópico externo. Posteriormente, se divide en dosmitades simétricas mediante un corte sagital. Por con-vención, en la mayoría de los bancos, la mitad derechase conservará en congelación, mientras que la mitad iz-quierda se fijará por inmersión en formaldehído tampo-nado al 10 % para estudio neuropatológico.
Existen diversas posibilidades de congelación del teji-do. El procedimiento más habitual, y más adecuado parala posterior obtención de muestras, es la realización delonchas coronales del hemisferio tras la separación deltronco, así como la realización de lonchas transversalesde tronco y sagitales de cerebelo. Las lonchas se conge-lan individualmente debidamente identificadas, median-te inmersión directa en nitrógeno líquido (protegiendo eltejido con papel de aluminio), aplicando las lonchas auna superficie metálica a baja temperatura, o por inmer-sión en isopentano a baja temperatura.
El tallado del hemiencéfalo fijado y la toma de muestrasse hace teniendo en cuenta las regiones cerebrales a lasque se aplican los criterios histológicos aquí recogidos.Se obtienen los siguientes bloques para inclusión en pa-rafina:
1. Córtex prefrontal dorso-lateral, incluyendo la circun-volución frontal media.
Este nivel es adecuado para estudiar algunos cambiosvasculares (signos de hipoxia-isquemia global, microin-fartos corticales, etc) por su localización en el límite delos territorios vasculares de las arterias cerebrales ante-
— 2 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
rior y media, y se corresponde con una de las regionesestablecidas en los criterios CERAD (4) para el diagnós-tico neuropatológico de la E. de Alzheimer.
2. Córtex cingular, incluyendo cuerpo calloso.
Permite estudiar el cuerpo calloso y la circunvolucióndel cíngulo, que forma parte del sistema límbico y seencuentra afectada en la E. de Alzheimer, y de formacaracterística en la Demencia con cuerpos de Lewy.
3. Sustancia blanca periventricular.
Permite estudiar cambios difusos en la sustancia blanca,y alteraciones localizadas en la región periventricular de-bidas a patología de pequeño vaso (leucoaraiosis).
4. N. estriado anterior, incluyendo cabeza del n. caudadoy putamen.
Dependiendo del tamaño del cerebro y el nivel seleccio-nado, permite un muestreo total del estriado anterior, in-cluyendo caudado, putamen y accumbens. Proporcionauna información indispensable tanto en patología neuro-degenerativa como en patología vascular.
5. N. estriado medio, incluyendo n. lenticular, así comon. basalis de Meynert .
Con este bloque se estudia el putamen en un segmentomás posterior, el globo pálido (regiones de interés en pa-tología neurodegenerativa y vascular), y permite incluirel n. basalis de Meynert, una región altamente sensiblea la neurodegeneración, particularmente en la E. de Alz-heimer y la Demencia con cuerpos de Lewy. Una alter-nativa es obtener uan muestra de n. basalis en un bloqueaislado, o incluirlo en un bloque con la amígdala.
6. Hipotálamo posterior, en el nivel de los cuerpos ma-milares.
Con este bloque se obtiene una buena representación delhipotálamo, incluyendo las paredes del III ventrículo,y los cuerpos mamilares. Estas regiones son importan-tes en algunas patologías neurodegenerativas y tóxicas(p.ej., encefalopatía alcohólica).
7. Córtex insular anterior.
El segmento anterior de la ínsula pertenece al sistema
límbico, y participa en la patología neurodegenerativa(Alzheimer, cuerpos de Lewy) que afecta más marcada-mente a este sistema.
8. Tálamo, incluyendo n. subtalámico y s. nigra rostral.
Si este bloque se toma del nivel adecuado (ver esquema),se obtiene una buena representación del tálamo, que per-mite estudiar sus regiones principales, del núcleo subta-lámico (esencial en algunas enfermedades neurodegene-rativas) y del segmento rostral de la sustancia nigra.
9. Corteza temporal externa, incluyendo circunvolucio-nes temporales superior y media.
Se trata de una de las regiones recomendadas por los cri-terios CERAD para el diagnóstico de la E. de Alzheimer.
10. Hipocampo anterior, incluyendo amígdala.
Este bloque permite examinar áreas de interés en enfer-medades neurodegenerativas, el complejo amigdalino, elcórtex periamigdalino, y el segmento anterior del hipo-campo.
11. Hipocampo medio-posterior.
Este es el nivel más adecuado para estudiar el hipocam-po. Es la estructura más sensible para estudiar cambiosde envejecimiento cerebral y lesiones iniciales de E. deAlzheimer, y se emplea para establecer el estadio evolu-tivo de esta enfermedad. También es un buen indicadorde isquemia-hipoxia global, especialmente el córtex delhipocampo (sectores CA1 y CA2).
12. Córtex parietal inferior.
Se trata también de una área establecida en los criteriosCERAD.
13. Circunvolución calcarina y córtex occipital asociati-vo vecino.
Permite estudiar el área visual primaria, y constituye uncomplemento indispensable para un estadiaje correctode la E. de Alzheimer.
14. Vermis cerebeloso superior.
— 3 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
El vermis es especialmente sensible a la atrofia asociadaa patología alcohólica y de otras causas.
15. Hemisferio cerebeloso, incluyendo corteza y n. den-tado.
Es posible incluir en un solo bloque varias folias cerebe-losas de la superficie superior del hemisferio cerebelosoy el n. dentado en profundidad. El córtex cerebeloso estambién especialmente sensible a la isquemia-hipoxia, yel núcleo dentado es una región diana en varias enferme-dades neurodegenerativas.
16. Mesencéfalo.
En el mesencéfalo es importante valorar la sustancia ni-gra, región diana en la E. de Parkinson y en otras enfer-medades neurodegenerativas, la sustancia gris periacue-ductal, los colículos y el núcleo rojo. El nivel de mesen-céfalo que se procesa para histología es el más rostralobtenido tras la separación del tronco.
17. Protuberancia (nivel medio).
En este nivel se puede valorar el locus coeruleus y otrosnúcleos del tegmento pontino, así como todas las estruc-turas de la base protuberancial.
18. Bulbo raquídeo (nivel medio).
En este nivel se valoran varios núcleos de pares cranea-les, tractos longitudinales, incluida la vía piramidal, ylos núcleos de la oliva inferior, que muestra patologíacon frecuencia en correlación con el cerebelo.
19. Médula cervical.
En ocasiones sólo es posible estudiar la unión bulbo-medular. Si se dispone de médula cervical, se toma elnivel más caudal incluido en la pieza. Si se ha extraídola médula completa se toman muestras a nivel cervical,dorsal y lumbar.
HALLAZGOS HISTOLÓGICOS Y CRITERIOSDIAGNÓSTICO
En todo proceso diagnóstico, establecer criterios supo-ne reducir considerablemente la información disponi-ble, seleccionando sólo aspectos relevantes. En cualquierpatología neurodegenerativa, los hallazgos histológicos
son muy ricos en términos de regiones cerebrales afecta-das, cambios neuronales y gliales, inclusiones celulares,depósitos extracelulares, etc. Sin embargo, el diagnósti-co de la mayoría de ellas se basa en pocos tipos lesio-nales y en pocas regiones cerebrales. Los criterios diag-nósticos de una entidad clínico-patológica deben ser:
SencillosTransferiblesValidadosVersátiles
Algunos ejemplos servirán para definir la línea, en oca-siones difusa y fluida, que separa los hallazgos histoló-gicos que configuran la descripción de un caso o de unapatología, de los criterios histológicos que nos permitenhacer el diagnóstico y la clasificación del tejido.
La espongiosis laminar cortical, p.ej., es un hallazgo fre-cuente en las demencias, pero tan inespecífico que ca-rece de valor diagnóstico. El cambio espongiforme delneuropilo (espongiosis) es característico de la mayoríade las enfermedades priónicas, y es un criterio diagnós-tico. También es muy frecuente observar este cambio enel córtex entorrinal de casos con Demencia con cuerposde Lewy, si bien no es en absoluto un criterio diagnósti-co para esta entidad (al contrario, es mejor considerarlocomo una posible fuente de error diagnóstico cuando eltejido es limitado, p. ej. en una biopsia cerebral).
La presencia de neuronas balonizadas es característicade varias enfermedades neurodegenerativas, y entre ellaslas taupatías y algunos subtipos de enfermedad prióni-ca. En el primer caso constituyen criterios diagnósticos,mientras que en el segundo son sólo hallazgos, por lodemás muy interesantes desde el punto de vista patogé-nico.
Por último, la angiopatía amiloide cerebral es muy ca-racterística de la E. de Alzheimer, pero no es específicade esta entidad. Si bien no se utiliza como criterio diag-nóstico, algunos grupos han observado que la presenciade extensa angiopatía amiloide se asocia significativa-mente a los criterios establecidos de EA (3).
No pretendemos, pues, aquí recoger descripciones com-pletas de la neuropatología de las demencias, sino fi-jarnos en aquellas lesiones que nos sirven hoy de crite-rio diagnóstico. Su historia reciente nos enseña que de-bemos considerarlos como relativamente transitorios, y
— 4 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
que seguramente veremos pronto surgir nuevas entida-des o nuevos subtipos en las que ya conocemos, basadasen datos que hoy forman parte de la descripción generalde los casos, o de una parte de ellos. Como nos enseñala cita de Heráclito, apoyados en lo que esperamos concriterio, podremos encontrar lo nuevo, variante o distintoque nos hará progresar en el conocimiento de una enti-dad.
ENFERMEDAD DE ALZHEIMER
La Enfermedad de Alzheimer (EA) constituye la demen-cia neurodegenerativa más frecuente. Afecta en Españaa unas 600.000 personas, en Europa a 5,5 millones, y enel mundo a unos 18 millones. El 53 % de los casos seencuentra en el rango de edad de 75 a 84 años, mien-tras que el 40 % tiene 85 años o más. La mayoría de loscasos se dan de forma esporádica, mientras que menosdel 5 % son de origen genético (EA familiar), debido amutaciones en los genes PS1, PS2 y APP. En los casosesporádicos el gen ApoE actúa como gen de susceptibi-lidad. Las proteínas patológicas asociadas en tejido ce-rebral a EA son la proteína tau (una proteína asociada alos microtúbulos) y el amiloide Abeta (que resulta de laescisión de la APP). Las principales lesiones histológi-cas de la EA, así como todos los criterios diagnósticosestablecidos hasta el momento, están relacionados conlos depósitos patológicos formados por estas proteínas.
Los criterios que incluimos a continuación se vienenaplicando a series de casos durante más de una década yhan servido de base al conocimiento molecular actual dela enfermedad.
Criterios CERAD
Fueron establecidos por el Consortium to Establish a Re-gistry for Alzheimer’s Disease (CERAD) y se publica-ron en 1991 (4). Surgieron de la necesidad de mejorarlos criterios utilizados hasta entonces, muy poco especí-ficos, basados en la cuantificación de placas seniles (5).Los objetivos del grupo eran: a) generar unos criteriosneuropatológicos más precisos y fiables para la EA, b)determinar el espectro neuropatológico de la EA, y c)establecer el tipo y la frecuencia de otras patologías quese dan de forma aislada o coexisten con la EA.
Se seleccionan 5 regiones anatómicas para estudio mi-croscópico (mínimo):
- circunvolución frontal media- circunvolución temporal superior y media- lóbulo parietal inferior- hipocampo y córtex entorrinal- mesencéfalo, incluyendo s. nigra
La evaluación se realiza en cortes de parafina de 6-8 mi-cras. Además de H/E y otras técnicas generales, se re-comienda una tinción argéntica como el Bielschowskymodificado. Se acepta tioflavina S (con luz UV) comoalternativa para visualizar placas y ONFs, y también Ro-jo Congo.
Los criterios CERAD se basan en la evaluación semi-cuantitativa de placas seniles neocorticales de tipo neu-rítico, esto es, aquellas que contienen neuritas engrosa-das positivas para tinción argéntica. (El protocolo tieneen cuenta la dificultad que puede existir en la identifica-ción de las placas de forma regular y consistente entredistintos laboratorios). La evaluación se realiza en trespasos.
Paso 1: Evaluación semicuantitativa de la frecuencia deplacas seniles y ONFs neocorticales en áreas de máxi-ma densidad. También se registra el grado de angiopatíaamiloide y la proporción de placas con núcleo amiloide.Se proporciona un esquema visual para definir las fre-cuencias leve, moderada y alta de placas (para tinción deplata y tioflavina).
Paso 2: Se obtiene un indice (score) de placas relativoa la edad, combinando la edad del paciente al fallecercon la medición semicuantitativa de placas en la regiónneocortical de afectación más intensa.
Paso 3: Este índice se integra con la información clíni-ca relativa a la presencia o ausencia de demencia paradeterminar el nivel de certeza del diagnóstico de EA.
(Ver Tabla 1)
El diagnóstico neuropatológico final se obtiene:
E. de Alzheimer definitiva: índice de placas C y demencia.E. de Alzheimer probable: índice de placas B y demencia.E. de Alzheimer posible: índice de placas A y demencia óíndice de placas B/C sin demencia
— 5 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
El protocolo recomienda además registrar la patologíavascular y la presencia de patología de E. de Parkinson.
Entre las críticas que ha recibido el protocolo CERAD,están la necesidad de una mayor precisión en las áreas detoma de muestra y la limitación de las técnicas, que noincluyen inmunohistoquímica (6), así como la dificultadpara basar la evaluación en una tinción estable y paradiferenciar entre densidad baja y moderada de placas (7).
Criterios NIA - Reagan
Estos nuevos criterios surgieron de la revisión, por partedel National Institute on Aging (NIA) y del Ronald andNancy Reagan Institute, de los criterios originales pro-puestos por el NIA (5), teniendo en cuenta la implanta-ción del CERAD y desarrollos posteriores, como el sis-tema de estadiaje de la patología neurofibrilar propuestopor Braak (8).
Directrices para el diagnóstico postmórtem de la E.de Alzheimer (9).
- La E. de Alzheimer es un entidad clínico-patológicaheterogénea. Por ello, sobre la base de los hallazgos pa-tológicos sólo se pueden hacer afirmaciones probabilis-tas sobre la presencia o ausencia de demencia, y los ha-llazgos postmórtem sólo pueden servir de base para unainferencia diagnóstica si hay evidencia de una demenciaprogresiva previa.
- Más de un proceso patológico puede contribuir a la de-mencia.
- Cualquier lesión de tipo Alzheimer debe considerarsecomo patológica, aunque parezca ser incidental.
Evaluación neuropatológica de la probabilidad deque la EA justifique la demencia:
- La probabilidad es alta cuando el cerebro postmórtemmuestra placas neuríticas (PN) y ovillos neurofibrilares(ONF) en el neocórtex: índice de PN frecuente segúnCERAD y Estadio V/VI de Braak y Braak.
- La probabilidad es intermedia si existe moderada den-sidad de PN y ovillos en regiones límbicas: CERAD mo-derado y estadio III/IV de Braak y Braak.
- La probabilidad es baja si la distribución e intensidadde PN y ovillos es más limitada: CERAD infrecuente yestadio I/II de Braak y Braak.
Recomendaciones específicas:
- Para el diagnóstico de rutina de la EA, se recomienda eluso de procedimientos semicuantitativos (CERAD) paraevaluar PN y ovillos. Se considera esencial, así mismo,la evaluación de ovillos en el hipocampo y el neocórtex.
- En el contexto de investigación de la EA, deben utili-zarse métodos de estadiaje topográfico (Braak) para es-tablecer la extensión de la patología neurofibrilar (PN,ovillos y hebras neuropílicas).
- Se recomiendan los protocolos CERAD para la fija-ción, el procesamiento y la tinción del tejido (Biels-chowsky, Gallyas, Tioflavina S). Así mismo, se reco-mienda la aplicación del estadiaje de Braak a cortes es-tándar de parafina, según procedimientos establecidos(10).
- Tras el examen macroscópico, deben tomarse las si-guientes muestras en los correspondientes cortes coro-nales del cerebro:
- Areas neocorticales: circunvolución temporal superior,lóbulo parietal inferior, córtex frontal medio, córtex oc-cipital, incluyendo córtex visual primario y córtex aso-ciativo.
* Formación del hipocampo en el nivel del núcleo geni-culado lateral.* Formación del hipocampo en el nivel del uncus, inclu-yendo córtex entorrinal.* Sustancia nigra y locus coeruleus.
Se recomienda opcionalmente la inclusión de otras áreasde estudio: tálamo, n. caudado, putamen, cerebelo, cór-tex motor, córtex cingulado, cuerpos mamilares y médu-la espinal.
Una aportación fundamental de los criterios NIA-
— 6 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Reagan fue la posibilidad de adaptar el estadiaje deBraak (8) al estudio NP convencional en parafina:
Cortes utilizados para la determinación del estadiode Braak:
1) hipocampo en el nivel del uncus,2) hipocampo en el nivel del c. geniculado lateral, y3) córtex occipital incluyendo c. calcarina y córtex aso-ciativo visual adyacente.
Áreas evaluadas:
- Córtex entorrinal-perirrinal (CE) medial al surco rinal,en elnivel de la amígdala.- Córtex del hipocampo (sector CA1) en el nivel del c. geni-culado lateral.- Córtex temporal inferior (CTI) lateral al surco colateral.- Córtex visual primario (CVP).- Córtex asociativo visual adyacente a la cisura calcarina.
Estadio 0: No se observan ovillos neurofibrilares(ONF) ni hebras neuropílicas (HN) en ninguna de las5 áreas.
Estadio I: Se observan escasos ONF y HN en la capaII del CE, y ninguno o muy ocasionales en CA1. No seidentifica patología neurofibrilar en neocórtex.
Estadio II: Se observan escasos o moderados ONF yHN en la capa II del CE, así como ocasionales o esca-sos ONF y HN en CA1. En CTI no se observa patologíaneurofibrilar, o es muy ocasional a este nivel. En córtexoccipital no se observa patología neurofibrilar.
Estadio III: En la capa II del CE se observan ONF yHN moderados o frecuentes, mientras que en CA1 sonescasos o moderados. En el CTI la patología neurofibri-lar es ocasional o escasa, y está ausente en las áreas decórtex occipital evaluadas.
Estadio IV: Se observan ONF y HN moderados o fre-cuentes en la capa II del CE y en CA1, así como pa-
tología neurofibrilar escasa o moderada en el CTI. Nose observa patología neurofibrilar en el córtex asociativovisual o es a lo sumo escasa. No se observa patología enel CVP.
Estadio V: La capa II del CE y el sector CA1 del hi-pocampo contienen frecuentes ONF Y HN. En el CTIse observan ONF y HN con intensidad moderada a fre-cuente. En cuanto al córtex occipital, la patología neu-rofibrilar es escasa o moderada en el córtex asociativo yesta ausente o es muy ocasional en el CVP.
Estadio VI: La patología neurofibrilar es frecuente enla capa II del CE en CA1 y en CTI. Se observan ONFmoderados o frecuentes en áreas asociativas visuales, yel CVP contiene ONF y HN ocasionales o escasos.
Para estudios de investigación, se han propuesto los cri-terios de la Universidad de Washington (11), basados enla evaluación cuantitativa de todas las placas seniles, conalta sensibilidad y especificidad.
Un último desarrollo, que refleja la práctica habitual des-de hace años en los laboratorios de neuropatología (12),ha sido la adaptación del estadiaje de Braak a las téc-nicas de inmunohistoquímica para tau en parafina (13).Una reciente evaluación de técnicas histológicas entrecentros europeos recomienda igualmente la aplicaciónde inmunotinción en parafina para la aplicación de loscriterios NIA-Reagan (14).
DEMENCIA CON CUERPOS DE LEWY YOTRAS SINUCLEINOPATÍAS
La evolución reciente de los criterios para el diagnósti-co neuropatológico de las enfermedades con patologíade tipo Lewy ha estado claramente marcada por los de-sarrollos técnicos en la tinción de las lesiones caracte-rísticas (inmunotinción para ubiquitina y posteriormentepara alfa-sinucleína) y por el modo adaptarlos a la fre-cuente combinación de patología de tipo Lewy y tipoAlzheimer. En las lesiones de tipo Lewy, el componenteprincipal es una forma agregada de una proteína presi-náptica, la alfa-sinucleína, que se encuentra asociada aotros componentes, como neurofilamentos fosforiladosy ubiquitina.
— 7 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Demencia en la enfermedad de Parkinson
Aunque la demencia en la E. de Parkinson puede tenerun sustrato neuropatológico diverso, como patología deAlzheimer, vascular o patología de tipo Lewy, se ha pro-puesto un estadiaje de la patología alfa-sinucleina (+)que permite determinar el grado de progresión de estapatología desde el tronco cerebral hasta el córtex (15),con buena correlación con el estado cognitivo de los pa-cientes con EP (16). Para el establecimiento de estos cri-terios se han excluido los casos que cumplen criteriosde Demencia con cuerpos de Lewy (ver más adelante) ylos casos con patología de tipo Alzheimer y patología deLewy cortical.
Hallazgos patológicos:
- Neuritas de Lewy (NL). Con alfa-sinucleína : puedenser gruesas, con forma de bastón o sacacorchos, puedenser cortas o de mayor longitud, como hebras; pueden te-ner conformación varicosa, y bifurcarse, terminando enensanchamientos en forma de lágrima.
- Cuerpos de Lewy (CL). Con alfa-sinucleína : puedenser esféricos o reniformes, de superficie lisa y tamañoy forma variables. Se pueden observar de forma aisladao en grupos entre los gránulos de lipofuchina o neuro-melanina. En ocasiones, se observan "cuerpos pálidos"menos definidos y con inmunorreactividad débil.
Estadio 1: Afectación del n. motor dorsal IX/X (neu-ronas no pigmentadas), y en ocasiones en la zona reticu-lar intermedia. En los casos con menos patología sólo seobservan NL, mientras que si la patología es más intensapredominan las NL sobre los CL.
Estadio 2: Afectación más intensa de las áreas afec-tadas en el estadio 1, con aparición de NL y Cl en losnúcleos del rafe caudal y en la formación reticular. Asímismo, se observa afectación del complejo coeruleus-subcoeruleus.
Estadio 3: Afectación de neuronas pigmentadas del n.motor dorsal IX/X y de la zona intermedia. Afectación
de un subconjunto de neuronas pigmentadas de la s. ni-gra (subnúcleos póstero-lateral y póstero-medial). Se ob-servan NL en el n. pedúnculopontino (porción compac-ta) y en los nn. magnocelulares del cerebro anterior basal(n. septal medial, n. intersticial de la banda diagonal y n.basal de Meynert). También en el n. túberomamilar hi-potalámico. En este estadio puede observarse un plexode NL en CA2 del córtex del hipocampo.
Estadio 4: Se observa intensa pérdida neuronal en lasregiones posteriores de la s. nigra compacta, y se observapigmento extracelular. Afectación de otros nn. mesen-cefálicos pigmentados: n. paranígrico y n. parabraquial.También nn. del rafe oral. Intensa afectación de los nn.magnocelulares del cerebro anterior basal. N. intersticialde la stria terminalis y la amígdala (nn. accesorios corti-cales y centrales) y el claustro. También en nn. específi-cos del tálamo. Daño intenso del n. olfatorio anterior. Unhallazgo constante en este estadio es el desarrollo de le-siones en el mesocórtex temporal anterior: capas super-ficiales, NL filiformes, capas inferiores, CL en neuronaspiramidales pequeñas o medianas.
Estadio 5: Afectación más intensa de todas las áreaspreviamente afectadas. En la s. nigra disminuye el nú-mero de NL y CL, y aumenta la cantidad de melaninaextraneuronal. Se observa igualmente pérdida de neuro-nas pigmentadas del n. motor dorsal del IX/X, la zonareticular intermedia, la formación reticular y el comple-jo coeruleus-subcoeruleus. Afectación intensa de áreasolfatorias. En el córtex del hipocampo afectación ini-cial de CA3 y CA1. Afectación neocortical, insulacórtexcingulado anterior y áreas prefrontales. En el neocórtex,predominio de afectación de neuronas piramidales infra-granulares.
Estadio 6: Afectación de la casi totalidad del neocór-tex: áreas premotoras, área motora primaria, áreas aso-ciativas sensitivas de primer orden y areas sensitivas pri-marias, con afectación relativamente leve.
Demencia con cuerpos de Lewy
Los criterios de consenso originales (17) fijaron una en-tidad clínico-patológica que venía describiéndose en la
— 8 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
literatura bajo diversas denominaciones. La reciente re-visión de estos criterios (18), permite, en su aspecto neu-ropatológico, una evaluación más sencilla (semicuantita-tiva) y reproducible de las lesiones y una co-evaluaciónde la patología de tipo Alzheimer y de tipo Lewy.
Existen dos interpretaciones en la literatura de la diferen-cia entre la E. de Parkinson con demencia y la demenciacon cuerpos de Lewy (DCL): 1) como dos fenotipos clí-nicos distintos dependientes de la secuencia de apariciónde la clínica, con una base neuropatológica común, y 2)como distintas presentaciones clínico-patológicas dentrode un espectro continuo de enfermedades con patologíade tipo Lewy. Esta última visión parece preferible desdeel punto de vista de investigación molecular y genética ypara el desarrollo terapéutico. Un caso se clasifica comoDCL si la demencia se desarrolla a la vez que el parkin-sonismo, y como E. de Parkinson con demencia cuandola demencia aparece en el contexto de una EP estableci-da (con un año de historia al menos).
Los criterios neuropatológicos originales se propusieroncon el fin de identificar una entidad clínico-patológicabien definida (DCL). Sin embargo, se ha comprobadoque hasta el 60 % de los casos de E. de Alzheimer cum-plen dichos criterios de DCL. Se han propuesto nuevasrecomendaciones para tener en cuenta conjuntamente laextensión de la patología de tipo Lewy y la patología detipo EA, con el fin de determinar el grado de certeza conque los hallazgos NP permiten explicar el síndrome clí-nico.
El procesamiento y toma de muestras del cerebro se rea-liza de acuerdo con el esquema del primer consenso (17).Regiones evaluadas (bloques):
Regiones neocorticales:
- Circunvolución frontal media en el surco frontal supe-rior, plano coronal inmediatamente anterior al polo tem-poral (Área de Brodmann 8/9).- Circunvolución temporal media, margen suoerior (sur-co temporal superior), plano coronal del cuerpo mamilar(Área de Brodmann 21).- Surco intraparietal (margen superior) del lóbulo parie-tal, plano coronal 1 cm posterior al polo posterior delesplenio del c. calloso (Área de Brodmann 40).
Regiones límbicas o paralímbicas:
- C. cingular anterior (Área de Brodmann 24).- C. transentorrinal (Área de Brodmann 29).
Regiones del tronco cerebral:
Sustancia nigra.Locus coeruleus.N. dorsal del vago.
Se realiza una evaluación semicuantitativa de la inten-sidad de la patología de tipo Lewy (leve, moderada, in-tensa y muy intensa), con un patrón similar al utilizadopara evaluar las PN y los ONF en CERAD. La patologíase identifica histológicamente median inmunostiquímicapara alfa-sinucleína.
En cada caso se determinará previamente la probabilidadde que la demencia pueda ser atribuida a la patología detipo EA de acuerdo con los criterios NIA-Reagan.
La patología de tipo Lewy se evalúa de forma semicuan-titativa en las siguientes áreas:
- N. motor dorsal IX-X.- Locus coeruleus.- S. nigra.- N. basal de Meynert.- Amígdala.- Córtex transentorrinal.- Córtex cingular.- Córtex temporal.- Córtex frontal.- Córtex parietal.
Teniendo en cuenta la intensidad de afectación de lasdistintas áreas, de acuerdo con una tabla de referencia,la patología de tipo Lewy del caso se clasifica como:
- Predominante en tronco cerebral.- Límbica (transicional).
Neocortical difusa.
Finalmente, después de aplicar los criterios NIA-Reaganpara E. de Alzheimer, el tipo de patología de Lewy se
— 9 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
cruza con la probabilidad obtenida de EA, de acuerdocon el siguiente esquema, para obtener la probabilidadde que la clínica del paciente pueda explicarse por la pa-tología de tipo Lewy (DCL).
(Ver Tabla 2)
Atrofia multisistémica
La atrofia multisistémica (AMS) es una enfermedad neu-rodegenerativa progresiva de causa desconocida, espo-rádica, que se presenta en dos subtipos clínicos princi-pales: la AMS-P (degeneración estriatonígrica (DEN)),con parkinsonismo dominante, y AMS-C (atrofia olivo-pontocerebelosa (AOPC)), con atrofia cerebelosa predo-minante.
Histológicamente, se caracteriza por la presencia de in-clusiones citoplásmicas gliales (ICG) en la oligodendro-glía, que constituye un criterio para el diagnóstico defi-nitivo de AMS (19). Las ICG son inclusiones argirófilas(se tiñen especialmente bien con la tinción de Gallyas),de conformación semilunar, oval o cónica, compuestaspor filamentos de 20-30 nm dispuestos en múltiples ca-pas. Contienen ubiquitina y antígenos citoesqueléticos.Inclusiones similares se observan en neuronas y astro-citos. En ocasiones se observan inclusiones neuronalesubiquitina-positivas, nucleares o citoplásmicas, simila-res a las observadas en la enfermedad de motoneuro-na, así como neuritas distróficas ubiquitina-positivas ytau-negativas. Estas inclusiones son positivas para alfa-sinucleína. Las ICG se distribuyen por los ganglios basa-les, el córtex motor primario y suplementario, la forma-ción reticular y el sistema pontocerebeloso (20). Hastael 10 % de los casos pueden presentar patología asocia-da de tipo Lewy.
Se observa pérdida neuronal, astrocitosis y microgliosisen el sistema estriatonígrico, más intensas en la zona la-teral dorsal del putamen caudal y en la porción lateral dela s. nigra pars compacta. El globo pálido y el n. subta-lámico también aparecen afectados. También se observapérdida neuronal en el hipotálamo, el bulbo raquídeo, n.arcuato y zona intermedia de la médula espinal, así comoen varios nn. precerebelosos y el n. ambiguo. La máximaintensidad de afectación se observa en el n. reticularlate-ral y el n. accesorio de la oliva inferior. En el cerebelo, seobserva afectación más intensa de las células de Purkin-
je en el vermis que en los hemisferios, con atrofia del n.de la oliva inferior, las fibras cerebelopontinas, y atrofiade la basis pontis.
Con el fin de categorizar adecuadamente los casos pa-ra investigación clínica o terapéutica, se ha propuesto elestadiaje por separado de la DEN y la AOPC (21). Enambos casos se definen 3 estadios basados en la evalua-ción semicuantitativa de la atrofia, la pérdida neuronal,la astrocitosis (ausente, mínima, leve, moderada e inten-sa), y del número de ICG positivas para alfa-sinucleína(ausentes, muy escasas, moderadas, y numerosas).
La evaluación de la DEN se realiza en:
- Putamen ventromedial y dorsolateral.- Caudado anterior y posterior.- Globo pálido medial y lateral.- Sustancia nigra compacta ventrolateral y dorsolateral.
La evaluación de la AOPC se realiza en:
- Base pontina.- Cerebelo (pérdida de células de Purkinje).- Oliva inferior.- Sustancia nigra compacta.
Cada caso queda así encuadrado en un estadio de DEN(I-III) y un estadio de AOPC (I-III).
DEMENCIAS VASCULARS Y DEMENCIASMIXTAS
La patología vascular, como sustrato neuropatológicoúnico de demencia (demencia vascular pura) o compar-tido con otras patologías, predominantemente de tipoAlzheimer (demencia mixta) ha sido la más resistenteal enunciado de criterios neuropatológicos de diagnós-tico (22). En series clínicas la demencia aparece comola segunda causa más frecuente de demencia (15-20 %de los casos), mientras que en series neuropatológicas lafrecuencia es menor como patología pura aunque muyvariable (3 % -13,5 %). Las lesions vasculares a menu-do coexisten con otras patologías, principalmente de ti-po Alzheimer. En las demencias mixtas, el efecto de lapatología vascular parece ser sumatorio, de modo queexiste una discordancia entre los índices de patología detipo Alzheimer (CERAD ó NIA-Reagan) y el grado dedéficit cognitivo (23, 24).
— 10 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Diversidad de etiologías:
- Hipertensión y ateroesclerosis- Enfermedad embólica- Vasculitis- S. Sneddon- Drogas de abuso- Tumores (linfoma intravascular)- CADASIL- Angiopatías amiloides familiares
Diversidad de hallazgos neuropatológicos:
- Macroangiopatía* Infartos extensos o múltiples* Infartos estratégicos
- Microangiopatía* Infartos lacunares (Ia, Ib, II y III)* Esclerosis arteriolar (lipohialinosis)* Expansión del espacio perivascular* Rarefacción y gliosis perivascular* Rarefacción difusa de la s. blanca(leucoencefalopatía subcortical /cambio de Binswanger)* Atrofia granular del córtex
- Lesiones por hipoperfusión* Esclerosis del hipocampo* Necrosis laminar cortical
- Hemorragia
Los criterios NINDS-AIREN (1993) de demencia vascu-lar definitiva están basados en criterios neuropatológicosnegativos (25):
- Demencia clínica probable- Histología de enfermedad vascular- Ausencia de otros criterios neuropatológicos de de-mencia- Descripción de las lesiones focales (infartos y lagunas)por territorios vasculares: número, volumen de tejido yestructuras anatómicas afectadas.- Descripción de la patología no oclusiva de pequeño va-so.
Patología vascular superficial- Ateroesclerosis -% oclusión (0, 20 %, 50 %, 80 %, 100 %)
* Arteria cerebral anterior
* Arteria cerebral media* Arteria posterior* Carótida* Arteria vertebral* Bifurcación carotídea* Tronco basilar
Otra patología vascular.- Lesiones Focales- Infartos, >10 mm Número, territorio y localización- Infartos lacunares, <10 mm Número y localización- Hemorragias Número, diámetro y localización
Patología microvascular (evaluación semicuantitativa:leve, moderada o severa)
- Sustancia gris* Esclerosis arteriolar* Expansión del espacio de Virchow-Robin* Gliosis y rarefacción perivascular
- Sustancia blanca* Esclerosis arteriolar* Expansión del espacio de Virchow-Robin* Pérdida difusa de mielina y gliosis de la s. blanca* Otros cambios microvasculares
- Centro semioval, en el nivel del estriado.- Lóbulo parietal, en el nivel del pulvinar.- Lóbulo occipital, en el polo occipital deventrículo lateral.
Sustancia gris:- Ganglios basales.- Tálamo.
Protuberancia:- Basis pontis.
CADASIL
En la arteriopatía autosómica dominante cerebral coninfartos subcorticales y leucoencefalopatía (CADASIL:Cerebral autosomal dominant arteriopathy with subcor-tical infarcts and leukoencephalopathy) el diagnósticohistológico desempeña un papel especial. La enferme-dad es debida a mutaciones en el gen NOTCH-3. Histo-lógicamente, se caracteriza por la presencia de un mate-
— 11 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
rial de depósito específico en la capa media vascular, os-miófilo en microscopía electrónica. El diagnóstico pue-de realizarse en biopsia cutánea con microscopía elec-trónica. Recientemente, se han desarrollado anticuerposmonoclonales frente al material de depósito que permi-ten detectarlo mediante inmunohistoquímica en biopsiascutáneas. Si bien el diagnóstico mediante inmunohisto-química en biopsia cutánea muestra una sensibilidad yespecificidad altas, la presencia de falsos negativos y fal-sos positivos aconseja confirmar el resultado en todos loscasos mediante análisis del DNA (29).
DEMENCIAS CON TAUPATÍA
Las taupatías constituyen un grupo de enfermedadesneurodegenerativas que tienen en común la presencia deinclusiones argirófilas citoplásmicas neuronales y glia-les compuestas por proteína tau fibrilar que, como en laE. de Alzheimer (tauopatía en sentido lato), muestra unpatrón anómalo de fosforilación. En el cerebro humanoadulto, el ensamblaje alternativo del RNAm (exones 2, 3y 10) de un gen situado en el cromosoma 17 da lugar a6 isoformas principales de tau. El ensamblaje alternativodel exon 10 genera isoformas de tau con 3 ó 4 repeticio-nes de los aminoácidos 30-32, que se conocen como taucon 3 repeticiones (tau 3R) y tau con 4 repeticiones (tau4R), respectivamente. En las distintas enfermedades deeste grupo, la acumulación de tau 3R y 4R en las neu-ronas y la glía muestra un patrón selectivo. En la E. deAlzheimer se observan los dos tipos, mientras que en laenfermedad de Pick (EP) predomina la tau 3R y exis-te depósito predominante de tau 4R en la degeneracióncórticobasal (DCB), la parálisis supranuclear progresiva(PSP) y la demencia con granos argirófilos (DGA).
Es muy importante tener en cuenta que en ocasiones lastaupatías, y especialmente las esporádicas, pueden pre-sentar fenotipos incompletos o combinados entre los quecorresponden a las entidades clínico-patológicas aquí in-cluidas.
Principales lesiones histológicas en las taupatías:
- Ovillos neurofibrilares (ONF), de conformación glo-boide o en llama, similares a los de la E. de Alzhei-mer, que se tiñen con inmunohistoquímica para tau y conBielschowsky o Gallyas.
- Pre-ovillos. Neuronas con inmunorreactividad cito-plásmica para tau, difusa y granular. No se detectan continciones argénticas.
- Hebras neuropílicas (HN), argirófilas y tau-positivas,de origen neuronal o glial. Se pueden observar especial-mente bien con Gallyas.
- Astrocitos en penacho. Es una lesión muy caracterís-tica, si bien no específica, de la PSP. Esta constitutidapor un penacho de fibras argirófilas y tau-positivas queconvergen en un pericarion astrocitario.
- Placas astrocitarias. Agrupación anular de procesos ce-lulares cortos que recuerda a una placa neurítica. Nocontienen amiloide, y los procesos son de origen astro-glial. Es la lesión histológica más característica de laDCB.
- Coiled bodies. Inclusiones citoplásmicas oligodendro-gliales tau positivas y argirófilas (tinción de Gallyas),más características de la PSP, si bien presentes en todaslas taupatías.
- Cuerpos de Pick. Inclusiones citoplásmicas argirófi-las redondeadas y bien delimitadas. Pueden observarsecon Bielschowsky, pero no con Gallyas. Muestran inmu-norreactividad para neurofilamentos fosoforilados, tau yubiquitina.
- Cuerpos córticobasales. Inclusiones homogéneas basó-filas y amorfas similares a ONF, características de la s.nigra en la DCB. Se pueden observar con H/E y son in-munorreactivas para tau.
- Granos argirófilos. Se trata de estructuras argirófilasfusiformes o en forma de coma que muestran inmuno-rreactividad para tau. Se detectan también con tinciónde Gallyas.
- Neuronas balonizadas (acromásicas). Se detectan me-diante H/E como neuronas de tamaño aumentado, núcleoexcéntrico y citoplasma homogéneo, débilmente eosi-nófilo. Muestran inmunorreactividad para NF y beta-cristalina, y débil para ubiquitina.
- Degeneración grumosa neuronal. Se trata de un tipo dedegeneración neuronal asociada a agrupaciones de ter-minales sinápticos patológicos en torno al cuerpo neuro-
— 12 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
nal. Se observa característicamente en el n. dentado encasos de PSP.
Parálisis supranuclear progresiva.
La PSP es una de las principales causas de parkinsonis-mo resistente a levodopa. Con frecuencia los pacientesdesarrollan cierto grado de trastorno cognitivo con pa-trón compatible con patología subcortical o frontal. Esuna enfermedad infrecuente, predominantemente espo-rádica, pero más frecuente que la DCB o la EPi.
En el examen externo lo más común es atrofia frontaly mesencefálica leve. El n. subtalámico puede estar cla-ramente atrófico. La s. nigra muestra despigmentaciónvariable, más acusada en la región ventrolateral. Se ob-serva discoloración grisácea del pedúnculo cerebelososuperior y del hilio del n. dentado.
Las lesiones características se identifican con tinción ar-géntica (preferiblemente Gallyas) e inmunotinción paratau. Se observa pérdida neuronal y astrocitosis, princi-palmente en áreas subcorticales, con máxima afectaciónde globo pálido, n. subtalámico, s. nigra y n. dentado delcerebelo. El estriado y el tálamo (núcleos ventrales ante-rior y lateral) pueden presentar astrocitosis. En el troncocerebral las áreas afectadas son los colículos superiores,la sustancia gris periacueductal, los nn. oculomotores, ellocus coeruleus, los núcleos pontinos, el tegmento pon-tino y bulbar y las olivas inferiores. En todas estas áreaspueden observarse ONF y HN, así comocoiled bodies,que también se observan en sustancia blanca subcortical.Los astrocitos en penacho son frecuentes en el estriadoy en el córtex. La patología cortical, que en ningún casoes intensa, es más marcada en córtex frontal, incluyendoc. precentral.
Estos hallazgos se han traducido en criterios diagnósti-cos semicuantitativos, válidos cuando existe historia clí-nica compatible con PSP (30):
- Presencia de alta densidad de ONF y HN en al menostres de las siguientes áreas: globo pálido, n. subtalámico,s. nigra y protuberancia, y- Presencia de densidad baja o alta de ONF y HN en almenos tres de las siguientes áreas: n. estriado, complejooculomotor, bulbo y n. dentado.
Raras veces es necesario aplicar estrictamente estos cri-terios. Cuando se plantea el diagnóstico diferencial his-tológico de la PSP con la DCB y los casos de E. de Alz-heimer con extensa patología subcortical, el predominiode afectación subcortical de la PSP y su característicoperfil de inclusiones gliales permite hacer el diagnósticosin dificultad.
Degeneración córticobasal.
La DCB es una enfermedad neurodegenerativa progresi-va infrecuente que se presenta como parkinsonismo asi-métrico con trastorno cognitivo (31).
La atrofia cortical, intensa y generalmente asimétrica,está centrada en las áreas perirrolándicas frontal y pa-rietal. Así mismo, se observa despigmentación de la s.nigra.
Las áreas corticales afectadas muestran desestructura-ción arquitectural, pérdida neuronal y gliosis. Las le-siones histológicas más características en el córtex sonlas neuronas balonizadas (acromásicas), la presencia deneuronas con positividad citoplásmica difusa para tau(preovillos) y placas astrocitarias. Las neuronas baloni-zadas son más frecuentes en las capas III, IV, V y VIcorticales. En las áreas de afectación subcortical puedendetectarse así mismo cuerpos corticobasales.
Histológicamente, en ocasiones no es fácil diferenciarentre la DCB, la PSP y la EPi. Sin embargo, en la ma-yoría de los casos el patrón macroscópico de afectacióncortical y la presencia abundante de neuronas baloniza-das y placas astrocitarias permite hacer el diagnósticosin dificultad.
Demencia con granos argirófilos.
Se interpreta en el momento actual como una enferme-dad infradiagnosticada, si bien su marcador histológico,la presencia de granos argirófilos, se ha detectado en el4 % de cerebros postmórtem estudiados (con o sin de-mencia) y en el 5-9 % de los cerebros con demencia. Asímismo, con frecuencia se encuentran asociados a otrastaupatías (EPi, DCB y PSP) (32).
— 13 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Los hallazgos macroscópicos son inespecíficos y en al-gunos casos se ha descrito atrofia del lóbulo temporalmedial. Histológicamente, se observan abundantes gra-nos argirófilos de distribución difusa en el neuropilo delcórtex entorrinal, la amígdala y el hipocampo. La sustan-cia blanca vecina contiene abundantes "coiled bodies", yse pueden detectar celulas balonizadas en áreas límbicas,especialmente en la amígdala.
Enfermedad de Pick.
En el momento actual la denominación de enfermedadde Pick está estrictamente limitada a casos que muestranatrofia lobar, cuerpos de Pick, neuronas balonizadas y unpatrón de isoformas de tau con predominio 3R (32).
El examen macroscópico del cerebro revela intensa atro-fia cortical, netamente delimitada y en ocasiones asimé-trica, de los lóbulos temporal y frontal, en su porciónanterior, la porción orbitaria del lóbulo frontal y la por-ción medial del lóbulo temporal. Los ventrículos apare-cen dilatados y en ocasiones se observa atrofia estriatal ypalidal y despigmentación de la s. nigra. El hipocampoy la amígdala suelen estar intensamente afectados, conabundantes cuerpos de Pick en el córtex del hipocam-po y en las células granulares del giro dentado. Tambiénpueden encontrarse cuerpos de Pick en el estriado y enel globo pálido. La s. nigra puede mostrar intensa pérdi-da neuronal, más marcada en la región rostral que en lacaudal.
Histológicamente, las áreas corticales afectadas mues-tran intensa pérdida neuronal y astrocitosis, con estatusespongioso. Las neuronas de las capas corticales supe-riores contienen cuerpos de Pick, mientras que en las ca-pas medias e inferiores se observan neuronas baloniza-das.
Demencia frontotemporal con parkinsonismo li-gada al cromosoma 17.
Los pacientes desarrollan una demencia frontotemporalcon marcado parkinsonismo y amiotrofia. La enferme-dad muestra herencia autosómica dominante y se asociaa mutaciones en el gen de la tau, en el cromosoma 17.
Los cambios macroscópicos son variables, desde intensaatrofia frontotemporal a casos con atrofia cortical asimé-trica similar a la observada en la DCB. La atrofia afectageneralmente a las porciones anterior e inferior de los ló-bulos temporales y a la circunvolución del cíngulo. Pue-de existir atrofia estriatal y despigmentación de la s. ni-gra.
Histológicamente, las áreas atróficas muestran pérdidaneuronal y gliosis. El córtex puede contener neuronasbalonizadas. Mediante tinción argéntica o inmunotin-ción para tau, se observan ONF, preovillos, hebras neu-ropílicas, cuerpos de Pick o inclusiones gliales tau posi-tivas.
El rasgo histológico más distintivo de las DFTP-17 esque el conjunto de inclusiones tau-positivas y su patrónde distribución no corresponde a ninguna de las taupatíasesporádicas.
DEMENCIAS FRONTOTEMPORALES SINTAUPATÍA
Con este grupo, enunciado sólo a efectos expositivos yde algoritmo diagnóstico, nos referimos estrictamente alas patologías que debemos considerar cuando no iden-tificamos inclusiones tau-positivas en un caso con pa-trón macro y microscópico de degeneración lobar fronto-temporal.
Con el término de demencia frontotemporal (DFT) sedesigna en el momento actual el principal síndrome clí-nico asociado a la degeneración de los lóbulos frontalesy temporales, y se caracteriza por cambios de la perso-nalidad y la conducta que progresan a apatía y mutismo.El sustrato neuropatológico de la DFT es heterogéneo, sibien los distintos subgrupos tiene en común la degenera-ción lobar frontotemporal y un inicio de la enfermedaden general antes de los 65 años.
El concepto actual de degeneración lobar frontotemporalcomo sustrato neuropatológico de las DFT surgió de ladefinición de criterios de consenso por parte de un grupode trabajo (33, 34). Los autores recomendaron un siste-ma de clasificación con 5 categorías neuropatológicas:
- Presencia de depósitos de tau insoluble con predominiode tau 3R.- Presencia de depósitos de tau insoluble con predominiode tau 4R.
— 14 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
- Presencia de inclusiones de tau insoluble 3R y 4R.- Ausencia de inclusiones tau-positivas y presenciade inclusiones de tipo enfermedad de motoneurona(ubiquitina-positivas) en el giro dentado del hipocampo.- Ausencia de inclusiones tau-positivas y ubiquitina po-sitivas.
Los tres primeros grupos de esta clasificación han sidoincluidos aquí dentro del apartado de taupatías. A con-tinuación se expone la visión actual de los dos gruposrestantes, que ahora forman cuatro grupos diferenciados(35, 36).
Degeneración lobar frontotemporal con inclu-siones ubiquitina-positivas.
Los pacientes descritos bajo esta entidad muestran clíni-ca de DFT, pero no presentan enfermedad de motoneuro-na clínica ni neuropatológica. Macroscópicamente, pre-sentan atrofia lobar frontal y temporal, y puede existirafectación estriatal y nígrica.
Histológicamente, los cambios se inician con pérdidaneuronal y astrocitosis en capas corticales superficiales(I-III), con espongiosis laminar a este nivel. Las neuro-nas restantes pueden mostrar intensa cromatolisis e in-clusiones eosinófilas pálidas. El rasgo histológico máscaracterístico es la presencia de inclusiones neuronalescitoplásmicas en las células granulares del giro denta-do, en la amígadala y en las capas superficiales neocor-ticales, y especialmente en la capa granular externa. Es-tas inclusiones son similares a los cuerpos de Pick perono son argirófilas ni inmunorreactivas para tau, y mues-tran positividad para ubiquitina. En las áreas afectadastambién se observan neuritas distróficas inmunorreacti-vas para ubiquitina. En ocasiones, las inclusiones son in-tranucleares.
Degeneración lobar frontotemporal asociada aenfermedad de motoneurona.
Los pacientes muestran un cuadro clínico de DFT, conclínica o datos neuropatológicos de enfermedad de mo-toneurona. Algunos casos son familiares. Se han descritocasos en los que no se observa patología de motoneuro-na, que se han considerado como presintomáticos. Ade-
más de la atrofia frontotemporal, como en otras degene-raciones lobares F-T, puede existir atrofia estriatal.
Histológicamente, se observan inclusiones ubiquitina-positivas superponibles a las observadas en la ELA enneuronas motoras. Se observan, así mismo, inclusionesextramotoras en el córtex frontal y temporal, idénticas alas descritas en la degeneración lobar F-T con inclusio-nes ubiquitina-positivas.
Degeneración lobar frontotemporal con inclu-siones de neurofilamentos.
Se trata de una entidad recientemente descrita (37), conun patrón macroscópico indistinguible de otras degene-raciones F-T, y atrofia estriatal. El rasgo específico deesta entidad es la presencia de inclusiones intraneurona-les inmunorreactivas para neurofilamentos y para alfa-internexina. Son negativas para tau, y débilmente posi-tivas para ubiquitina. Con H/E aparecen como peque-ñas inclusiones redondeadas y basófilas, de conforma-ción variable, entre 5 y 20 micras de diámetro.
Demencia sin rasgos histológicos específicos.
Esta entidad, también denominada Degeneración lobarsin inclusiones neuronales simplemente Degeneraciónlobar frontotemporal aparece en las series publicadas porexclusión, es morfológicamente superponible a las ante-riores, salvo por la ausencia de inclusiones de cualquiertipo y patrón de inmunorreactividad. Algunos autoreshan atribuido la falta de inmunorreactividad a ubiquitinaa problemas técnicos, e interpretan la mayoría de estoscasos como formas de degeneración F-T ubiquitina- po-sitivas (38).
ENFERMEDADES POR PRIONES
De acuerdo con los criterios de vigilancia epidemiológi-ca, el diagnóstico definitivo de las enfermedades huma-nas por priones exige confirmación neuropatológica, enla gran mayoría de los casos mediante estudio postmór-tem. La única excepción a esta regla la constituyen loscasos de enfermedad de origen genético por mutaciónpatogénica detectadaintra vitam, así como los raros ca-sos de E. de Creutzfeldt-Jakob esporádica (ECJe) diag-
— 15 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
nosticados por biopsia cerebral. El diagnóstico postmór-tem puede realizarse en la mayoría de los casos sobreescaso material (un bloque de tejido de córtex cerebral,estriado o cerebelo incluido en parafina) estudiado contécnicas adecuadas (incluyendo inmunohistoquímica pa-ra PrP) (39). Sin embargo, la correcta clasificación de uncaso de enfermedad priónica humana requiere un proto-colo combinado de estudio neuropatológico y molecu-lar. El estudio neuropatológico revela el perfil fenotípi-co morfológico (regiones del SNC afectadas, patrón deafectación regional y patrón de depósito de PrP). El estu-dio genético permite detectar posibles mutaciones en elgen PRNP, y proporciona el status del polimorfismo delcodón 129. El estudio molecular de tejido cerebral no fi-jado detecta el tipo o los tipos de PrPres implicados encada caso. La clasificación final de un caso requerirá laintegración de toda la información clínica, morfológicay molecular.
La ECJ esporádica, que representa el 85 % de todas lasenfermedades priónicas humanas, constituye su paradig-ma neuropatológico. El examen macroscópico no revelahallazgos específicos. Con frecuencia se observa atro-fia global, con afectación cortical evidente, más frecuen-te en el lóbulo occipital, o afectación macroscópica es-triatal, talámica, o cerebelosa (con atrofia, generalmentemás marcada del vermis). Es notable la preservación re-lativa del hipocampo. Un estudio adecuado en parafinaincluye al menos la toma de muestras de córtex (todoslos lóbulos), hipocampo, estriado, tálamo, tronco cere-bral (mesencéfalo, protuberancia y bulbo) y médula cer-vical. Los bloques de tejido fijado, previamente a su in-clusión en parafina, deben ser descontaminados median-te inmersión en ácido fórmico al 98 % durante una hora,con agitación suave, manteniéndolos posteriormente enformaldehído al 10 % durante 48 horas.
Un estudio histológico convencional con tinción dehematoxilina-eosina (H/E) permite observar la tríada ca-racterística de cambio espongiforme del neuropilo, pér-dida neuronal y gliosis (astrogliosis e incremento y acti-vación microglial). Laespongiosisde la ECJ se caracte-riza por vacuolas neuropílicas de límites netos y conteni-do óptimamente claro, con frecuencia de pequeño tama-ño y en ocasiones grandes y confluentes que, cuando sonperineuronales, producen una muesca en el cuerpo neu-ronal. Este cambio muestra una distribución transcorti-cal o afecta a las capas corticales profundas. Se observacon gran intensidad igualmente en córtex entorrinal, nú-cleos de la base, incluyendo tálamo, y capa molecular
cerebelosa. En el IFL la espongiosis es mínima o estáausente. En la forma "panencefalopática" de ECJ, se ob-serva extensa afectación de la sustancia blanca.
La pérdida neuronal, evidente por lo general con H/E,puede hacerse más patente mediante tinción de Nissl.En algunos casos pueden observarse neuronas baloni-zadas, que muestran inmunorreactividad para neurofila-mentos. Tinciones para mucopolisacáridos, como el PASy el azul Alcián, tiñen los depósitos de PrP en forma deplaca de "tipo kuru". Las tinciones para amiloide, comoel rojo Congo y la tioflavina S, pueden contribuir a de-mostrar los depósitos de PrP en forma de amiloide (pla-cas), aunque el tratamiento con ácido fórmico elimina latinción de rojo Congo.
La astrocitosises igualmente evidente con H/E, y confrecuencia en áreas de gran pérdida neuronal se observanabundantes astrocitos hipertróficos de aspecto gemisto-cítico. La inmunotinción para proteína acídica gliofibri-lar (PAGF) puede poner de manifiesto la astrocitosis conespecial expresividad. Igualmente, laactivación micro-glial, visible con H/E, puede demostrarse con inmuno-tinción para CD 68.
El depósito masivo de PrPresen tejido cerebral, no siem-pre paralelo a los cambios histológicos descritos, se de-tecta mediante tinción inmunohistoquímica. La interpre-tación de los resultados debe tener en cuenta igualmenteel protocolo de recuperación antigénica y revelado em-pleado en el laboratorio. También la tinción de Gallyasrevela algunos de los depósitos de PrP. Mediante inmu-notinción para PrP se observan los siguientes patronesde depósito1:
* Sináptico: intenso punteado fino de distribución difu-sa en el neuropilo, que puede asociarse a depósitos másgroseros e irregulares. Con frecuencia la corteza cerebe-losa muestra punteado fino de la molecular y depósitosgroseros en la granular.* Perivacuolar: depósitos parcheados y groseros en tor-no a vacuolas de tamaño mediano o grande.* Perineuronal: intenso depósito en torno a neuronas,que dibuja con nitidez el cuerpo neuronal y los segmen-tos proximales de las prolongaciones celulares.* Tipo placa: los depósitos de tipo placa, que son losúnicos que se extienden a la sustancia blanca, son másfrecuentes que lasplacas de tipo kuru(que se detec-tan sin inmunohistoquímica y son positivas para técnicasde amiloide). Lasplacas unicéntricasse dan en la ECJ
— 16 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
y la E. de Gerstmann-Sträusssler-Scheinker (GSS), lasplacas multicéntricasso características de la GSS, y lasplacas floridasson típicas de la variante de ECJ (ECJv),aunque se han descrito en algunos casos de ECJy portransplante de duramadre.
Las enfermedades priónicas humanas admitían clási-camente una doble clasificación clínico-patológica (E.de Creutzfeldt-Jakob, Insomnio familiar letal, Síndromede Gerstmann-Sträusssler-Scheinker, Kuru) y etiológica(esporádicas, genéticas y transmitidas). Desde hace unosaños, se añade a estas una tercera clasificación molecu-lar, basada en la mutación del gen PRNP (enfermeda-des genéticas), el polimorfismo del codón 129 del mis-mo gen y el tipo o los tipos de PrPres detectados en te-jido cerebral. A continuación se refieren las principalescaracterísticas microscópicas de cada una de estas enti-dades clínico-patológico-moleculares.
Enfermedad de Creutzfeldt-Jakob esporádica (EC-Je): Se han descrito 6 subtipos moleculares con hallaz-gos morfológicos homogéneos (40). Cada tipo se desig-na por el polimorfismo del codón 129 (p.ej., MM parametionina/metionina) seguido del tipo de PrPres detec-tado en tejido por Western blot (1 ó 2 para la ECJ espo-rádica).
- MM1 ó MV1(ECJ mioclónica o de Heidenhein). Es elsubtipo más frecuente (72 % de ECJe). Presenta cambiosneuropatológicos típicos en córtex cerebral con distribu-ción transcortical (máxima intensidad en córtex occipitalen 47 % de los casos), estriado, tálamo medial y cerebe-lo. El patrón de depósito de PrP es sináptico.- VV2 (ECJ cerebelosa o atáxica). Corresponde al 15 %de los casos. Se observa espongiosis en estructuras lím-bicas (a menudo con distribución laminar), estriado, tála-mo, hipotálamo, cerebelo y núcleos del tronco. El córtexoccipital está afectado en casos de larga evolución. Elpatrón de depósito de PrP es de tipo placa, perineuronaly sináptico.- MV2 (ECJ cerebelosa o atáxica, variante con placa detipo kuru). Corresponde al 8 % de ECJe. Histología simi-lar a VV2, pero con depósitos en forma de placas de tipokuru unicéntricas en la granular cerebelosa y, en casosde larga evolución, también en córtex, estriado y tálamo.- MM2-cortical. Corresponde aproximadamente al 2 %de ECJe. El perfil lesional es similar al de MM1, si biensin espongiosis en el cerebelo. Además, el patrón de es-
pongiosis es de vacuola grande, más intensa en córtexcerebral y estriado. El patrón de depósito de PrP es amenudo perivacuolar.- MM2-talámico. Es indistinguible de lo que se ha deno-minado insomnio familiar esporádico, y superponible alque se describe aquí en el apartado de IFL.- VV1. Corresponde al 1 % de los casos y se da en in-dividuos más jóvenes. La afectación es principalmentecórtico-estriatal, con relativa preservación del lóbulo oc-cipital y del cerebelo. Se observan frecuentes células ba-lonizadas en el córtex. El patrón inmunohistoquímico dePrP es de tipo sináptico e intensidad débil.
En el 20-25 % de los casos, el tejido cerebral contienesimultáneamente PrPres de tipos 1 y 2, y recientemen-te, se ha sugerido que la combinación de tipos puede sernotablemente más frecuente (41). El tipo 1 predomina enlos casos MM, y el tipo 2 en los casos VV. El perfil pa-tológico depende del tipo de PrPres dominante, aunquepuede ser mixto.
Se ha propuesto una clasificación análoga basada en 6 ti-pos de PrPres obtenidos mediante una técnica diferente(42), que muestra una alta correspondencia con la ex-puesta y que incluye un subtipo MM6 nuevo. Histológi-camente, este subtipo se caracteriza por extensa espon-giosis confluente en córtex y estriado, con afectación ce-rebelosa focal. El patrón inmunohistoquímico de PrP essináptico y perivacuolar.
Enfermedad de Creutzfeldt-Jakob familiar (ECJf).Al igual que el fenotipo clínico, el perfil neuropatoló-gico de las distintas mutaciones conocidas depende delhaplotipo que contiene la mutación y del alelo corres-pondiente en el codón 129 (43,44).
* E200K-129M. Se trata de la mutación más frecuen-te. El patrón neuropatológico es muy similar al subtipoMM1 de ECJe, y de hecho se detecta PrPres de tipo 1 entejido cerebral.* E200K-129V. El perfil neuropatológico es similar alsubtipo VV2 de ECJe, y se detecta PrPres de tipo 2 entejido cerebral.* D178N-129V. El perfil lesional es similar al subtipoVV1 de ECJe, y en tejido cerebral se detecta PrPres detipo 1.* V210I-129M. Se trata probablemente de una mutacióncon baja penetrancia. Los 7 casos descritos mostraron
— 17 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
afectación de córtex cerebral, estriado, tálamo y cerebe-lo.* T183A-129M. En los casos descritos se ha observadoafectación cortical y estriatal. No se observa depósito dePrP cortical, y en el estriado es de tipo placa.
Los haplotipos M232S-129 y V180J-129M han sido des-critos sólo en Japón. La mayoría de otros haplotipos ra-ros se asemejan morfológicamente al subtipo MM1 deECJe. Finalmente, las mutaciones por inserciones de oc-tapéptidos muestran fenotipos altamente variables quedependen de la longitud de la inserción, y las insercionesde 8-9 octapéptidos muestran un fenotipo de GSS.
Enfermedad de Creutzfeldt-Jakob iatrogénica(ECJy). En los casos de ECJy por transplante de du-ramadre, los hallazgos neuropatológicos son similaresa los de la ECJe. Se ha descrito un caso con la formapanencefalopática, y un subtipo que presenta plascasfloridas similares a las de la ECJv. Los casos debidosa hormonas hipofisarias muestran, además, intensaafectación cerebelosa con placas de tipo kuru e intensosdepósitos de PrP en la médula espinal.
Variante de Enfermedad de Creutzfeldt-Jakob(ECJv). Los casos de ECJv se caracterizan morfoló-gicamente por extensa afectación de corteza cerebral,especialmente occipital, cerebelosa, estriatal y talámica,más marcada en núcleos talámicos posteriores (pulvi-nar), con intensa gliosis a este nivel. Mediante técnicasconvencionales se observan placas floridas en cortezacerebral y cerebelosa. Estas placas están formadaspor un núcleo eosinofílico denso del que parten fibrasmás pálidas en sentido radial, y rodeado por un halode espongiosis. La inmunotinción para PrP demuestraestas placas, así como otros grupos de placas de menortamaño, y un depósito de tipo perineuronal en estriadoy cerebelo. También pueden detectarse depósitos dePrP mediante inmunohistoquímica en tejido linfoide detodo el organismo. Tanto en el SNC como en el tejidolinfoide se detecta por Western blot PrPres de tipo 2B(45).
Insomnio familiar letal (IFL). Mientras que, como sevio anteriormente, el haplotipo D178N-129V da lugar aun fenotipo de ECJ y a un tipo 1 de PrPres, el haplotipo
D178N-129M da lugar al fenotipo de IFL. La caracte-rística histológica invariable del IFL es la atrofia de losnúcleos ventral anterior, medio-dorsal y pulvinar del tá-lamo, con intensa astrogliosis, en ausencia de espongio-sis. Así mismo, se observa atrofia de las olivas inferioresy, en lo casos de larga evolución, espongiosis focal encorteza entorrinal y neocórtex. No se observan depósitosde PrP mediante inmunohistoquímica.
Enfermedad de Gerstmann-Sträussler-Scheinker(GSS). En algunas mutaciones, como la P102L, elfenotipo está condicionado por el codón 129 y por elcodón 219. Si bien existe una gran diversidad fenotípicaentre distintas mutaciones, y aun en un mismo haploti-po, la principal característica morfológica del GSS esla presencia de depósitos de PrPres en forma de placasde amiloide en el córtex y en cerebelo, típicamentemulticéntricas, con frecuente afectación estriatal ydel tracto piramidal. Así mismo, se observan ovillosneurofibrilares en P105L-129L, H187R-129V, F198-129V, D202N-129V, Q217R-129V y Y145STOP-129M.En esta última mutación se desarrolla una angiopatíaamiloide cerebral por PrP.
Kuru. Histológicamente, el kuru se caracteriza por es-pongiosis cortical laminar de distribución límbica conintensa afectación estriatal y cerebelosa. El cerebelocontiene frecuentes placas típicas que se tiñen con PAS.Las placas son de tamaño variable, con un diámetro má-ximo de 30, y se tiñen mediante inmunohistoquímica pa-ra PrP, que revela así mismo depósitos de tipo sinápticoy perineuronal.
REFERENCIAS
1. Guidelines on autopsy practice. Report of a working groupof the Royal College of Pathologists. The Royal College ofPathologists, 2002.
2. VV.AA. Enfermedad de Creutzfeldt-Jakob y otras Encefalo-patías Espongiformes Transmisibles Humanas. Guía de in-formación y recomendaciones para personal sanitario. Mi-nisterio de Sanidad y Consumo, Madrid, 2003.
3. Neuropathology Group of the Medical Research CouncilCognitive Function and Ageing Study (MRC CFAS). Pat-hological correlates of late-onset dementia in a multicentre,community-based population in England and Wales. Lancet2001; 357: 169-75.
4. Mirra SS. Heyman A, McKeel D, Sumi SM, Crain BJ,Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg Let al. The Consortium to Establish a registry for Alzhei-mer’s Disease (CERAD). Part II. Standardization of the neu-
— 18 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
ropathologic assessment of Alzheimer’s disease. Neurology1991; 41: 479-86.
5. Khachaturian ZS. Diagnosis of Alzheimer’s Disease. ArchNeurol 1985; 42: 1097-1104.
6. McLean CA, Beyreuther K, Masters CL. Commentaryon the Consensus Recommendations for the Post MortemDiagnosis of Alzheimer’s Disease. Neurobiol Dis, 1997; 18(4) Suppl 1: S89-S90.
7. Murayama S, Saito Y. Neuropathological diagnostic criteriafor Alzheimer’s disease. Neuropathology 2004; 24: 254-60.
8. Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239-59.
9. Hyman BT, Trojanowski JQ. Editorial on recommendationsfor the postmortem diagnosis of Alzheimer Disease from theNational Institute on Aging and the Reagan Institute Wor-king Group on diagnostic criteria for the neuropathologicalassessment of Alzheimer Disease. J Neuropathol Exp Neu-rol 1997; 56: 1095-7.
10. Newell K, Hyman BT, Growdon JH, Hedley-Whyte ET.Evaluation of NIA-Reagan Institute criteria for the neuro-pathological diagnosis of Alzheimer’s disease. J Neuropat-hol Exp Neurol 1997; 56: 593
11. McKeel Jr. DW, Price JL, Miller P, Grant EA, Xiong C, BergL, Morris JC. Neuropathologic criteria for diagnosing Alz-heimer Disease in persons with pure dementia of Alzheimertype.
12. Dickson DW. Required techniques and useful molecularmarkers in the neuropathologic diagnosis of neurodegene-rative diseases. Acta Neuropathol 2005; 109: 14-24.
13. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tre-dici K. Staging of Alzheimer disease-associated neurofibri-llary pathology using paraffin sections and immunocytoche-mistry. Acta Neuropathol 2006; July (Epub ahead of print).
14. Alafuzoff I, Pikkarainen M, Al-Sarraj S, Arzberger T, BellJ, Bugiani I et al. Interlaboratory comparison of AlzheimerDisease-related lesions: A study of the BrainNet EuropeConsortium. J Neuropathol Exp Neurol 2006; 65: 740-57.
15. Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen SteurENH, Braak E. Staging of brain pathology related to spora-dic Parkinson’s disease. Neurobiol Aging, 2003 : 197-211.
16. Braak H, Rüb U, Jansen Steur ENH, Del Tredici K, de VosRAI. Cognitive status correlates with neuropathologic stagein Parkinson disease. Neurology 2005; 64: 1404-10.
17. McKeith IG, Galasko D, Kosaka K et al. Consensus guide-lines for the clinical and pathological diagnosis of dementiawith Lewy bodies (DLB): report of the consortium on DLBinternational workshop. Neurology 1996; 47: 1113-24.
18. McKeith IG, Dickson DW, Lowe J et al. Diagnosis and ma-nagement of dementia with Lewy bodies. Third report of theDLB consortium. Neurology 2005; 65: 1863-72.
19. Lantos PL. The definition of multiple system atrophy: areview of recent developments. J Neuropathol Exp Neurol1998;57:1099-1111.
20. Wenning GK, Colosimo C, Geser F, Poewe W. Multiple sys-tem atrophy. Lancet 2004; 3: 93-103.
21. Jellinger KA, Seppi K, Wenning GK. Grading of neuropat-hology in multiple system atrophy: proposal for a novel sca-le. Mov Disord 2005; 20 (Suppl1): S29-36.
22. Morris JC. Dementia update 2005. Alzheimer Dis Assoc Di-sord 2005; 19: 100-117.
23. Snowdon DA, Greiner LH, Mortimer JA et al. Brain infarc-
tion an the clinical expression of Alzheimer disease. TheNun study. JAMA 1997; 277: 813-17.
24. Petrovitch H, Webster Ross G, Steinhorn SC, Abbott RD etal. AD lesions and infarcts in demented and non-dementedJapanese-American men. Ann Neurol 2005; 57: 98-103.
25. Roman GC, Tatemichi TK, Erkinjuntti T et al. Vascular de-mentia: diagnostic criteria for research studies. Report ofthe NINDS-AIREN International Workshop. Neurology, 43:250-60.
26. Esiri MM, Wilcock GK, Morris JH. Neuropathological as-sessment of the lesions of significance in vascular dementia.J Neurol Neurosurg Psychiatr 1997; 63 : 749-53.
27. Morris JH, Kalimo H, Viitanen M. Vascular dementias. En:The neuropathology of dementia. Eds. : M. Esiri, VMY Leeand JQ Trojanowski. CUP, Cambridge 2004.
28. Heyman A, Fillenbaum GG, Welsh-Bohmer KA, GearingM, Mirra SS, Mohs RC, Peterson BL, Pieper CF. Cerebralinfarcts in patients with autopsy-proven Alzheimer’s disea-se: CERAD, part XVIII Consortium to Establish a Registryfor Alzheimer’s Disease. Neurology 1998; 51: 159-62.
29. Lesnik Oberstein SAJ, van Duinen SG, van den Boom R, etal. Evaluation of NOTCH3 immunostaining in CADASIL.Acta Neuropathol 2003; 106: 107-11.
30. Hauw JJ, Daniel SE, Dickson D, Horoupian DS, JellingerH et al. Preliminary NINDS neuropathological criteria forSteele-Richardson-Olszewski syndrome. Neurology 1994;44: 20115-9.
31. Mahapatra RK, Edwards MJ, Schott JM, Bhatia KP. Corti-cobasal degeneration. Lancet Neurol 2004; 3: 736-43.
32. Dickson DW. Sporadic tauopathies: Pick’s disease, corti-cobasal degeneration, progressive supranuclear palsy andargyrophilic grain disease. En: The neuropathology of de-mentia. Eds. : M. Esiri, VMY Lee and JQ Trojanowski.CUP, Cambridge 2004.
33. McKhann GM, Albert MS, Grossman M, Miller B, DicksonD, Trokanosvski JQ. Clinical and pathologic diagnosis offrontotemporal dementia. Report of a work group on fron-totemporal dementia and Pick’s disease. Arch Neurol 2001;58: 1803-9.
34. Trojanowski JQ, Dickson D. Update on the neruopatholo-gical diagnosis of frontotemporal dementias. J NeuropatholExp Neurol 2001 ; 60 : 1123-6.
35. Mott RT, Dickson DW, Trojanowski JQ, Zhukareva V, LeeVM et al. Neuropathologic, biochemical, and molecularcharacterization of the frontotemporal dementias. J Neuro-pathol Exp Neurol. 2005 ; 64 : 420-8.
36. Shi J, Shaw CL, Du Plessis D, Richardson AMT, Bailey KL,Julien C et al. Histopathological changes underlying fronto-temporal lobar degeneration with clinicopathological corre-lation. Acta Neuropathol 2005; 110; 501-12.
37. Cairns NJ, Perry RH, Jaros E, Burn D, McKeith IG et al.Patients with a novel neurofilamentopathy: dementia withneurofilament inclusions. Neurosci Lett 341: 177-180.
38. Mackenzie IR, Shi J, Shaw CL, Duplessis DL, Snowden JS,Mann DM. Dementia lacking distinctive histology (DLDH)revisited. Acta Neuropathol 2006 (Epub ahead of print).
39. Budka H, Head MW, Ironside JW, Gambetti P, Parchi P,Zeidler M. Tagliavini F. Sporadic Creutzfeldt-Jakob Disea-se. In: Neurodegeneration: the molecular pathology of de-mentia and movement disorders. Ed.: D Dickson. ISN Neu-ropath Press, Basel, 2003.
— 19 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
40. Parchi P, Giese A, Capellari S, Brown P, Schulz-SchaefferW, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, PoserS, Rojiani A, Streichemberger N, Julien J, Vital C, Ghet-ti B, Gambetti P, Kretzschmar H. Classification of sporadicCreutzfeldt-Jakob disease based on molecular and phenoty-pic analysis of 300 subjects. Ann Neurol 1999;46:224-33.
41. Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A,Aguzzi A. Coexistence of multiple PrPSC types in indivi-duals with Creutzfeldt-Jakob disease. Lancet Neurol 2005;4: 805-14.
42. Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, BudkaH, Ironside JW, Collinge J. Molecular classification of spo-radic Creutzfeldt-Jakob disease. Brain 2003;126:1333-46.
43. Parchi P, Capellari S, Chen SG, Gambetti P. FamilialCreutzfeldt-Jakob Disease. In: Neurodegenration: the mo-lecular pathology of dementia and movement disorders. Ed.:D Dickson. ISN Neuropath Press, Basel, 2003.
44. Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadicand familial CJD: classification and characterisation. BritishMedical Bulletin 2003; 66: 213-239.
45. Head MW, Bunn TJR, Bishop MT, Mcloughlin V, Lowrie S,McKimmie CS et al. Prion protein heterogeneity in sporadicbut not variant Creutzfeldt-Jakob disease: U.K. cases 1991-2002. Ann Neurol 2004; 55: 851-9.
— 20 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
ICONOGRAFÍA
Tabla 1.-
Tabla 2.-
— 21 —����� ��������������� � ��� �� ��
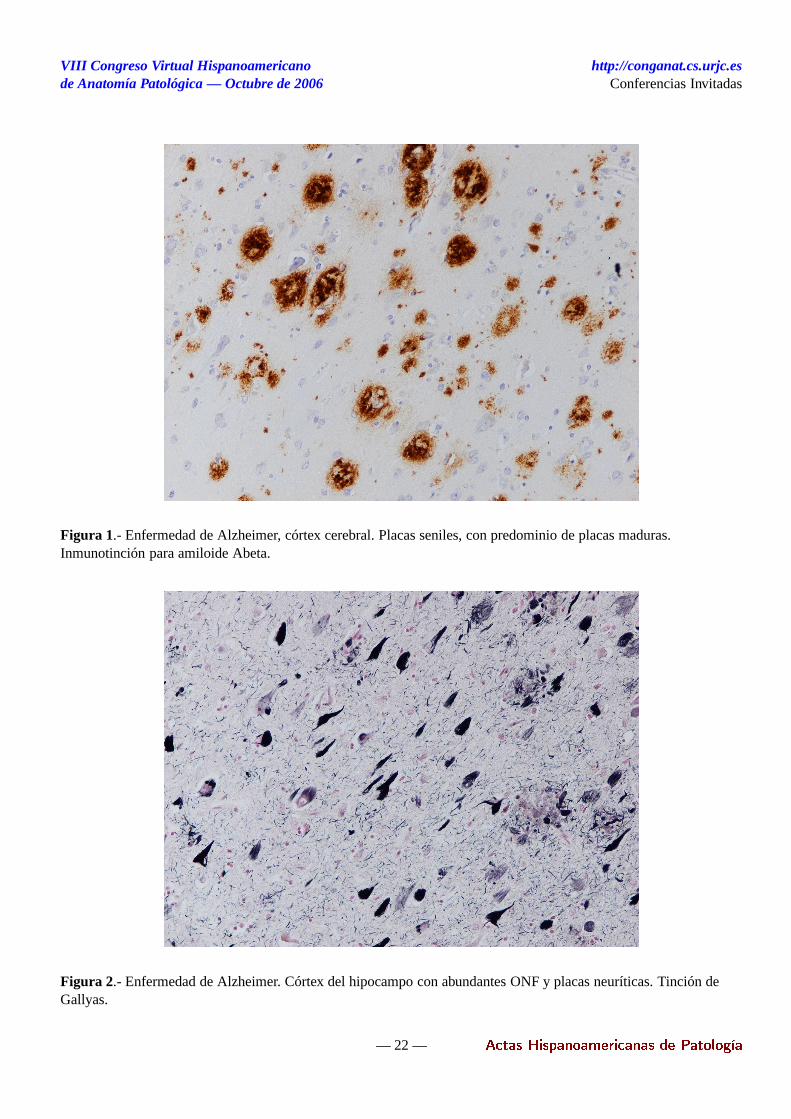
VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Figura 1.- Enfermedad de Alzheimer, córtex cerebral. Placas seniles, con predominio de placas maduras.Inmunotinción para amiloide Abeta.
Figura 2.- Enfermedad de Alzheimer. Córtex del hipocampo con abundantes ONF y placas neuríticas. Tinción deGallyas.
— 22 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Figura 3.- Enfermedad de Alzheimer. Córtex del hipocampo con abundantes ONF y placas neuríticas. Inmunotinciónpara tau (AT8).
Figura 4.- Enfermedad de Parkinson. Sustancia nigra. Cuerpo de Lewy. Hematoxilina-eosina.
— 23 —����� ��������������� � ��� �� ��
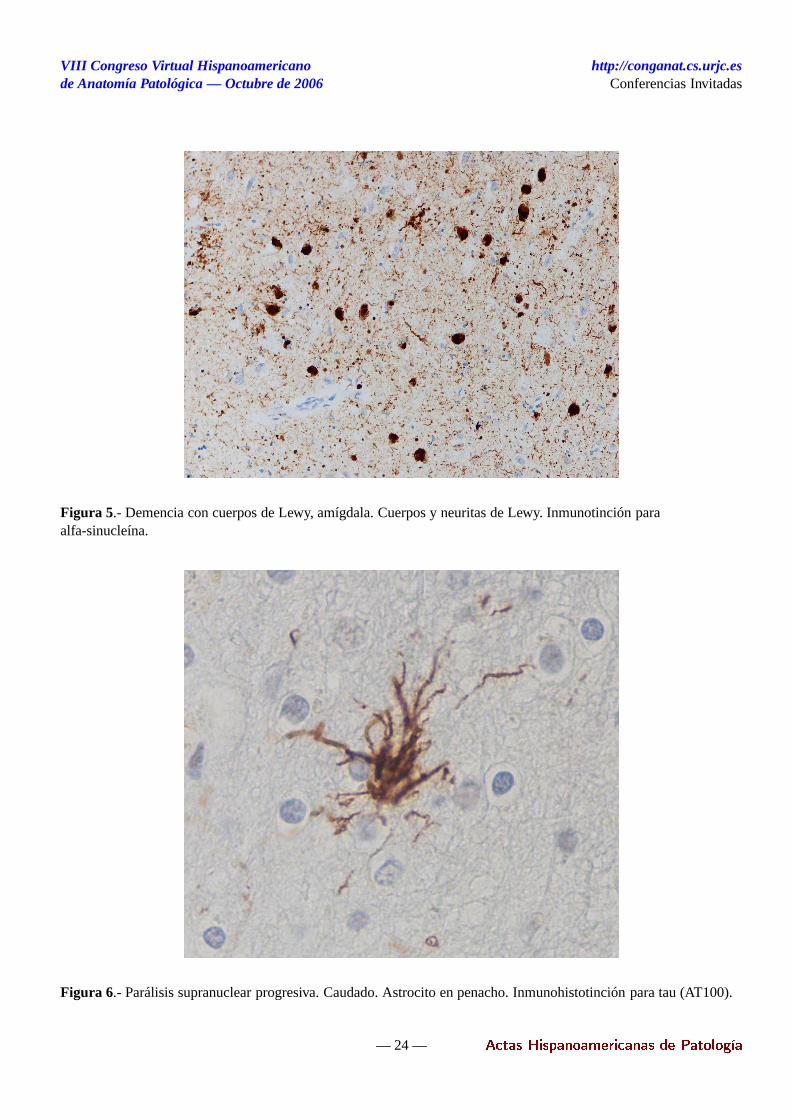
VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Figura 5.- Demencia con cuerpos de Lewy, amígdala. Cuerpos y neuritas de Lewy. Inmunotinción paraalfa-sinucleína.
Figura 6.- Parálisis supranuclear progresiva. Caudado. Astrocitoen penacho. Inmunohistotinción para tau (AT100).
— 24 —����� �������� ������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Figura 7.- Degeneración córticobasal. Neurona balonizada. Inmunotinción para neurofilamentos.
Figura 8.- Demencia frontotemporal con taupatía. Inclusiones tau-positivas en el giro dentado del hipocampo.Inmunotinción para tau.
— 25 —����� ��������������� � ��� �� ��

VIII Congreso Virtual Hispanoamericanode Anatomía Patológica — Octubre de 2006
http://conganat.cs.urjc.esConferencias Invitadas
Figura 9.- Degeneración lobar frontotemporal con inclusiones ubiquitina-positivas. Giro dentado del hipocampo.Inmunotinción para ubiquitina.
Figura 10.- Enfermedad de Creutzfeldt-Jakob esporádica. Córtex cerebeloso. Extenso depósito de proteína priónica,con patrón sináptico. Inmunotinción para PrP.
— 26 —����� �������� ������� � ��� �� ��


















