Guia de Practicas de Patologia General 2012 EVD
74
GUIA DE PRACTICAS DE PATOLOGÍA GENERAL PA 2320 TERCER° Año de Medicina LIMA – PERÚ 2012
-
Upload
jorge-pilco-inga -
Category
Documents
-
view
879 -
download
11
Transcript of Guia de Practicas de Patologia General 2012 EVD

GUIA DE PRACTICAS DE PATOLOGÍA GENERAL
PA 2320
TERCER° Año de Medicina
LIMA – PERÚ 2012

2
PRÁCTICAS DE MICROSCOPIA
Se llevarán a cabo en el laboratorio de prácticas de Histología de martes a viernes en el horario de 10:00 am a
12:00 m. y excepcionalmente de 12:00 a 01:30 p.m. Para esto se ha dividido a los alumnos en 4 grupos. El grupo
3 realizará prácticas los días martes, el grupo 1 los miércoles, el grupo 2 los jueves y el grupo 4 los viernes.
Práctica 1: PATOLOGIA CELULAR I
Lámina 1. Apoptosis
Lámina 2. Esteatosis hepática
Lámina 3. Amiloidosis renal
Lámina 4. Metaplasia intestinal
Lámina 5. Hiperplasia endometrial
18 – 21 de Setiembre
Dra. Bravo
Dr. Cáceres
Práctica 2: PATOLOGIA CELULAR II
Lámina 6. Hipertrofia muscular
Lámina 7. Atrofia endometrial
Lámina 8. Necrosis coagulativa
Lámina 9. Necrosis caseosa
Lámina 10. Necrosis grasa enzimática
25 – 28 de setiembre
Dra. Bravo
Dr. Nava
Práctica 3: INFLAMACIÓN AGUDA Y CRÓNICA
Lámina 11. Gastritis aguda y crónica
Lámina 12. Apendicitis
Lámina 13. Neumonía bacteriana
Lámina 14. Pielonefritis crónica
Lámina 15. Colecistitis crónica reagudizada
02 – 05 de octubre
Dr. Cok
Dr. Oscco
Práctica 4: TRANSTORNOS HEMODINÁMICOS
Lámina 16. Infarto pulmonar
Lámina 17. Trombo de la orejuela
Lámina 18. Edema pulmonar
Lámina 19. Infarto de miocardio
Lámina 20. Congestión vascular esplénica
09 – 12 de octubre
Dr. Chian
Dra. Del Castillo
Práctica 5: TRANSTORNOS GENÉTICOS
Lámina 21. Esferocitosis hereditaria
Lámina 22. Anemia de células falciformes
Lámina 23. Hemocromatosis
Lámina 24. Epidermólisis ampollar
Lámina 25. Enfermedad de Gaucher
16 – 19 de octubre
Dra. Bravo
Dr. Nava
Práctica 6: NEOPLASIAS BENIGNAS
Lámina 26. Leiomioma
Lámina 27. Neurofibroma
Lámina 28. Hemangioma capilar
Lámina 29. Adenoma velloso
Lámina 30. Meningioma
30 de octubre – 02 de noviembre
Dr. Cok
Dra. Del Castillo
Práctica 7: NEOPLASIAS MALIGNAS
Lámina 31. Carcinoma epidermoide
Lámina 32. Adenocarcinoma de ovario
Lámina 33. Linfoma de Burkitt
Lámina 34. Linfoma de Hodgkin
Lámina 35. Seminoma
06 – 09 de noviembre
Dra. Bravo
Dr. Nava

3
Práctica 8: ENFERMEDADES INFECCIOSAS
Lámina 36. Papilomavirus
Lámina 37. Citomegalovirus
Lámina 38. Helicobacter pylori
Lámina 39. Tuberculosis
Lámina 40. Leishmaniasis
13 – 16 de noviembre
Dr. Cáceres
Dra. Tapia
Práctica 9: ENFERMEDADES AMBIENTALES Y NUTRICIONALES
Lámina 41. Mesotelioma
Lámina 42. Hepatitis alcohólica
Lámina 43. Anemia aplásica
Lámina 44. Neumoconiosis
Lámina 45. Queratosis actínica
20 – 23 de noviembre
Dra. Del Castillo
Dr. Nava
Práctica 10: INMUNOPATOLOGÍA
Lámina 46. Pénfigo vulgar
Lámina 47. Glomerulonefritis lúpica
Lámina 48. Lepra lepromatosa
Lámina 49. Poliarteritis nodosa
Lámina 50. Nódulo reumatoide
27 – 30 de noviembre
Dr. Cok
Dr. Oscco
4
PRÁCTICA N° 1: PATOLOGIA CELULAR I
Lámina 1. APOPTOSIS
Es un proceso de muerte celular auto inducida en el que la membrana plasmática de las células permanece
intacta, mientras el ADN de la célula y las proteínas nucleares y citoplasmáticas se degradan. Este proceso
ocurre en situaciones fisiológicas o patológicas y se activa cuando se trata de eliminar células que ya no son
necesarias, células potencialmente dañinas o células que han sufrido un daño irreparable en su ADN. La
apoptosis es muy activa durante la embriogénesis, la evolución hormonal sexual, el ciclo de crecimiento de todos
los epitelios y el desarrollo del sistema inmunológico. También se produce en respuesta a diferentes estímulos
fisicoquímicos y biológicos que dañan irreversiblemente a las células. En ocasiones la apoptosis y la necrosis
coexisten y pueden compartir ciertas características y mecanismos.
Historia clínica. Paciente varón de 25 años en tratamiento con Trimetropin-Sulfametoxazol por una infección
intestinal que al tercer día de tratamiento presenta una erupción eritematosa maculopapular, con formación de
vesículas, de distribución asimétrica en codos, rodillas y porción extensora de antebrazos. La erupción persistió
por 7 días y remitió dejando áreas hiperpigmentadas en la piel afectada.
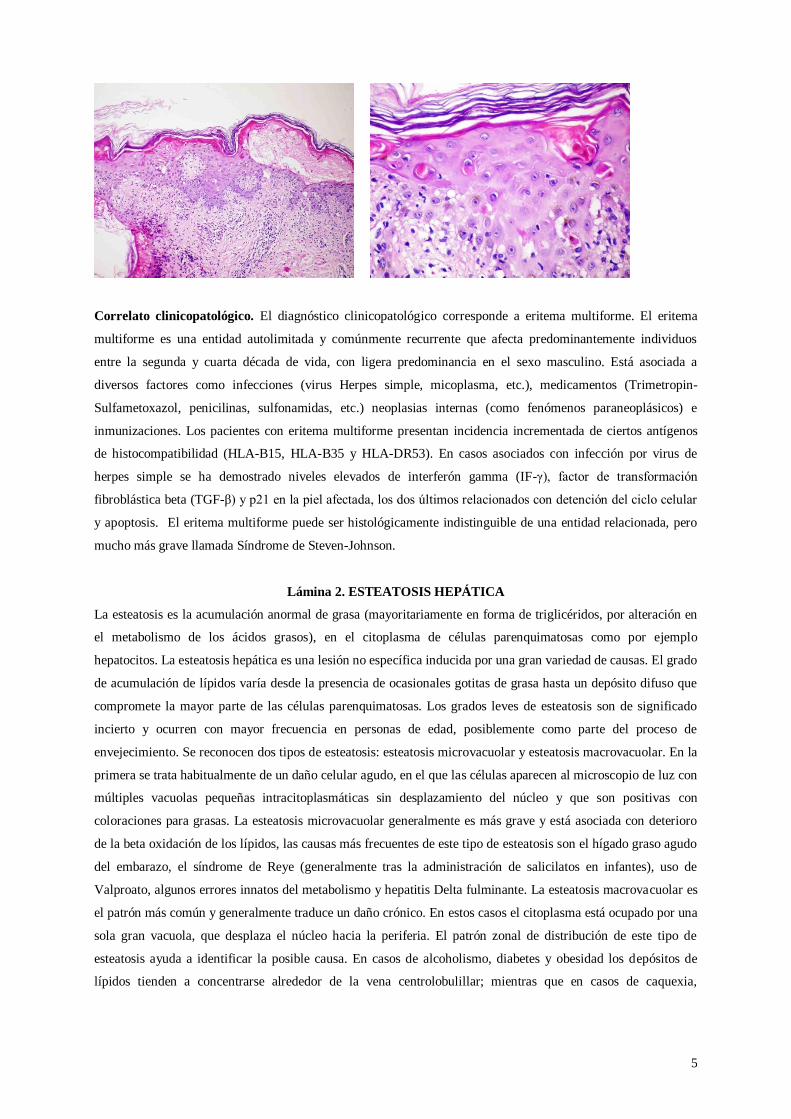
Lámina. Se observa degeneración hidrópica de la capa basal (formación de vacuolas a nivel de la unión entre la
epidermis y la dermis), numerosos queratinocitos apoptóticos, formación de vesículas y un infiltrado
inflamatorio superficial linfohistiocítico. Los queratinocitos apoptóticos son redondeados, intensamente
eosinofílicos y a menudo anucleados, aunque se pueden observar ocasionales figuras picnóticas residuales.
Lámina 1. Apoptosis
Lámina 2. Esteatosis hepática
Lámina 3. Amiloidosis renal
Lámina 4. Metaplasia intestinal
Lámina 5. Hiperplasia endometrial
5
Correlato clinicopatológico. El diagnóstico clinicopatológico corresponde a eritema multiforme. El eritema
multiforme es una entidad autolimitada y comúnmente recurrente que afecta predominantemente individuos
entre la segunda y cuarta década de vida, con ligera predominancia en el sexo masculino. Está asociada a
diversos factores como infecciones (virus Herpes simple, micoplasma, etc.), medicamentos (Trimetropin-
Sulfametoxazol, penicilinas, sulfonamidas, etc.) neoplasias internas (como fenómenos paraneoplásicos) e
inmunizaciones. Los pacientes con eritema multiforme presentan incidencia incrementada de ciertos antígenos
de histocompatibilidad (HLA-B15, HLA-B35 y HLA-DR53). En casos asociados con infección por virus de
herpes simple se ha demostrado niveles elevados de interferón gamma (IF-γ), factor de transformación
fibroblástica beta (TGF-β) y p21 en la piel afectada, los dos últimos relacionados con detención del ciclo celular
y apoptosis. El eritema multiforme puede ser histológicamente indistinguible de una entidad relacionada, pero
mucho más grave llamada Síndrome de Steven-Johnson.
Lámina 2. ESTEATOSIS HEPÁTICA
La esteatosis es la acumulación anormal de grasa (mayoritariamente en forma de triglicéridos, por alteración en
el metabolismo de los ácidos grasos), en el citoplasma de células parenquimatosas como por ejemplo
hepatocitos. La esteatosis hepática es una lesión no específica inducida por una gran variedad de causas. El grado
de acumulación de lípidos varía desde la presencia de ocasionales gotitas de grasa hasta un depósito difuso que
compromete la mayor parte de las células parenquimatosas. Los grados leves de esteatosis son de significado
incierto y ocurren con mayor frecuencia en personas de edad, posiblemente como parte del proceso de
envejecimiento. Se reconocen dos tipos de esteatosis: esteatosis microvacuolar y esteatosis macrovacuolar. En la
primera se trata habitualmente de un daño celular agudo, en el que las células aparecen al microscopio de luz con
múltiples vacuolas pequeñas intracitoplasmáticas sin desplazamiento del núcleo y que son positivas con
coloraciones para grasas. La esteatosis microvacuolar generalmente es más grave y está asociada con deterioro
de la beta oxidación de los lípidos, las causas más frecuentes de este tipo de esteatosis son el hígado graso agudo
del embarazo, el síndrome de Reye (generalmente tras la administración de salicilatos en infantes), uso de
Valproato, algunos errores innatos del metabolismo y hepatitis Delta fulminante. La esteatosis macrovacuolar es
el patrón más común y generalmente traduce un daño crónico. En estos casos el citoplasma está ocupado por una
sola gran vacuola, que desplaza el núcleo hacia la periferia. El patrón zonal de distribución de este tipo de
esteatosis ayuda a identificar la posible causa. En casos de alcoholismo, diabetes y obesidad los depósitos de
lípidos tienden a concentrarse alrededor de la vena centrolobulillar; mientras que en casos de caquexia,

6
malnutrición proteica, SIDA, nutrición parenteral total, envenenamiento por fósforo y terapia esteroidea el
patrón predominante suele ser periportal. A veces la esteatosis es tan severa que toma un patrón panlobular. La
esteatosis microvacuolar o de gota pequeña puede ser difícil de identificar y a veces es necesario hacer
coloraciones histoquímicas (Oil red O ó Sudán negro) en biopsias por congelación de tejido hepático. Los
aspectos clínicos de la esteatosis incluyen hepatomegalia y niveles elevados de aminotransferasas séricas,
fosfatasa alcalina y/o gammaglutamil transferasa hepática.
Historia clínica: Paciente mujer de 34 años con historia de malnutrición crónica. Durante el curso de una
colecistectomía por cálculos vesiculares se observa un hígado de color amarillento y se toma una biopsia en
cuña.
Lámina: Sección histológica de hígado que muestra preservación de la estructura lobulillar. A nivel periportal se
observa vacuolas dentro del citoplasma de los hepatocitos, que representan lípidos disueltos durante el
procesamiento del tejido. Algunas células muestran desplazamiento del núcleo hacia la periferia debido al
acumulo graso. Los espacios porta presentan leve infiltrado inflamatorio crónico.
Lámina 3. AMILOIDOSIS RENAL
El término amiloidosis designa a un grupo de condiciones caracterizadas por depósitos extracelulares de
proteínas fibrilares que tiene una configuración beta en láminas plegadas al ser analizadas mediante difracción
con rayos X. La insolubilidad y resistencia relativa a la digestión proteolítica permite que el amiloide se
acumule en los tejidos y altere las funciones de este, causando destrucción de órganos vitales y produciendo la

7
muerte. A pesar que los depósitos tienen una apariencia y características tintoriales similares la amiloidosis
incluye a un grupo de depósitos de naturaleza distinta en el que se conocen de acuerdo a la secuencia
aminoacídica de la fibrilla amiloidea, tres formas bioquímicas mayores (AA, AL y ATTR) y varias menores, que
se depositan por mecanismos patogénicos diferentes. En cada tipo se ha encontrado en el suero sanguíneo de los
enfermos una proteína precursora, que tiene la misma secuencia aminoacídica que la del amiloide del caso
depositado en los tejidos. La amiloidosis de tipo AL está relacionada con depósitos de cadenas ligeras completas
de inmunoglobulinas, de los fragmentos del extremo NH2 de las cadenas ligeras o ambos y generalmente ocurre
en el contexto de discrasias de células plasmáticas. La amiloidosis de tipo AA generalmente es secundaria y
suele estar asociada con enfermedades inflamatorias crónicas como tuberculosis, osteomielitis, bronquiectasias,
úlceras por decúbito, lepra, enfermedad de Crohn y artritis reumatoide. La amiloidosis de tipo ATTR es de tipo
familiar y en este caso generalmente las fibras de amiloide están constituidas por la proteína transtiretina mutada.
No existe forma de diferenciar el tipo de amiloidosis desde el punto de vista clínico.
El depósito de amiloide es extracelular y corresponde siempre a una condición patológica. La amiloidosis, no
rara vez es progresiva y letal. El daño local principal que produce la infiltración amiloidea es la atrofia. Estos
depósitos pueden ser localizados, o generalizados. Los órganos con amiloidosis presentan aumento de volumen a
pesar de la atrofia parenquimatosa que produce la infiltración amiloidea; la consistencia está aumentada, es firme
y elástica.
El reconocimiento histológico se hace mediante la tinción con Rojo de Congo, colorante que tiñe al amiloide de
color pardo rojizo. El tejido así teñido debe ser examinado con microscopia de luz polarizada, con la que el
amiloide presenta característicamente un color verde manzana.
Los depósitos amiloideos pueden desaparecer cuando la causa que los produce es tratada. Esto puede suceder,
por ejemplo, en tuberculosis, parasitosis y osteomielitis crónicas tratadas y en tumores extirpados relacionados
con amiloidosis generalizadas. La reabsorción es más rápida en el hígado y bazo (donde toma meses), en cambio
es más lenta en el glomérulo (donde demora años).
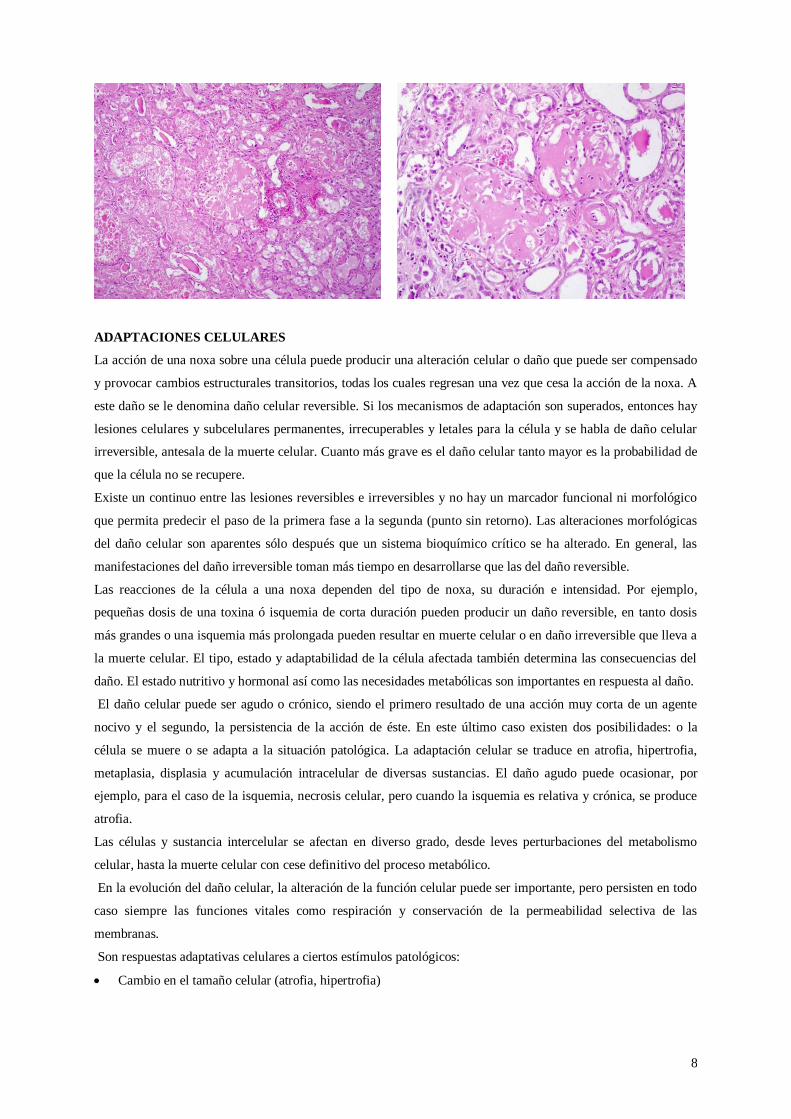
Historia clínica: Paciente varón de 54 años con antecedente de tuberculosis pulmonar que tiene un episodio
reciente de síndrome nefrótico (compromiso renal con proteinuria severa, edema de miembros inferiores).
Lámina: Fragmento de corteza y médula renal. A nivel de la corteza se identifican glomérulos, túbulis y vasos.
Se observa depósitos de un material amorfo, acelular, de color rosado pálido a nivel del mesangio glomerular
que en algunos casos llega a obliterar la luz capilar. Hay glomérulos convertidos en masas globulosas debido a la
gran cantidad de acumulación de material amiloide. También se observa depósitos a nivel de las paredes
vasculares. Los túbulos renales no tienen depósitos, pero están atróficos (la luz tubular está dilatada, el epitelio
aplanado con pérdida de las vellosidades y contienen cilindros hialinos).
8
ADAPTACIONES CELULARES
La acción de una noxa sobre una célula puede producir una alteración celular o daño que puede ser compensado
y provocar cambios estructurales transitorios, todas los cuales regresan una vez que cesa la acción de la noxa. A
este daño se le denomina daño celular reversible. Si los mecanismos de adaptación son superados, entonces hay
lesiones celulares y subcelulares permanentes, irrecuperables y letales para la célula y se habla de daño celular
irreversible, antesala de la muerte celular. Cuanto más grave es el daño celular tanto mayor es la probabilidad de
que la célula no se recupere.
Existe un continuo entre las lesiones reversibles e irreversibles y no hay un marcador funcional ni morfológico
que permita predecir el paso de la primera fase a la segunda (punto sin retorno). Las alteraciones morfológicas
del daño celular son aparentes sólo después que un sistema bioquímico crítico se ha alterado. En general, las
manifestaciones del daño irreversible toman más tiempo en desarrollarse que las del daño reversible.
Las reacciones de la célula a una noxa dependen del tipo de noxa, su duración e intensidad. Por ejemplo,
pequeñas dosis de una toxina ó isquemia de corta duración pueden producir un daño reversible, en tanto dosis
más grandes o una isquemia más prolongada pueden resultar en muerte celular o en daño irreversible que lleva a
la muerte celular. El tipo, estado y adaptabilidad de la célula afectada también determina las consecuencias del
daño. El estado nutritivo y hormonal así como las necesidades metabólicas son importantes en respuesta al daño.
El daño celular puede ser agudo o crónico, siendo el primero resultado de una acción muy corta de un agente
nocivo y el segundo, la persistencia de la acción de éste. En este último caso existen dos posibilidades: o la
célula se muere o se adapta a la situación patológica. La adaptación celular se traduce en atrofia, hipertrofia,
metaplasia, displasia y acumulación intracelular de diversas sustancias. El daño agudo puede ocasionar, por
ejemplo, para el caso de la isquemia, necrosis celular, pero cuando la isquemia es relativa y crónica, se produce
atrofia.
Las células y sustancia intercelular se afectan en diverso grado, desde leves perturbaciones del metabolismo
celular, hasta la muerte celular con cese definitivo del proceso metabólico.
En la evolución del daño celular, la alteración de la función celular puede ser importante, pero persisten en todo
caso siempre las funciones vitales como respiración y conservación de la permeabilidad selectiva de las
membranas.
Son respuestas adaptativas celulares a ciertos estímulos patológicos:
Cambio en el tamaño celular (atrofia, hipertrofia)
9
Cambio del número de células (hiperplasia)
Cambio de la diferenciación (metaplasia)
Lámina 4. METAPLASIA INTESTINAL
Se denomina metaplasia a la transformación o reemplazo de un tejido adulto por otro de la misma clase. Por
ejemplo, la metaplasia pavimentosa o escamosa del epitelio respiratorio de los bronquios en fumadores o en
bronquios que drenan cavernas tuberculosas; metaplasia pavimentosa del cuello uterino; metaplasia pavimentosa
del urotelio en la litiasis; metaplasia intestinal de la mucosa gástrica en la gastritis crónica; metaplasia intestinal
en la mucosa del esófago («esófago de Barrett»); metaplasia glandular en epitelio de la vejiga; metaplasia ósea
en cartílagos de la laringe; metaplasia ósea en cicatrices. La importancia de la metaplasia radica en que en
algunos casos, especialmente en la metaplasia intestinal en el esófago se asocia con un riesgo elevado de
displasia y de progresión a adenocarcinoma de esófago.
Los factores más a menudo relacionados son:
Irritación. Ejemplos: La litiasis en la metaplasia pavimentosa del urotelio. El reflujo de jugo gástrico ácido
en la metaplasia glandular del esófago (Esófago de Barrett).
Sustancias químicas. Ejemplos: Metaplasia escamosa en el epitelio bronquial en los fumadores; la
administración de nitrosamina en animales de experimentación determina la diferenciación de las células
acinares pancreáticas hacia hepatocitos.
Estrógenos: Un exceso de estrógenos produce metaplasia pavimentosa del epitelio cilíndrico del endocérvix
y de la próstata.
Déficit de vitamina A: Produce metaplasia pavimentosa en epitelios de la nariz, bronquios, epitelios
secretores de glándulas lacrimales y salivales.
Patogenia: Lo habitual es que la metaplasia se realice a partir de una célula indiferenciada o poco diferenciada
capaz de multiplicarse. A partir de ella se generan células con diferente diferenciación.
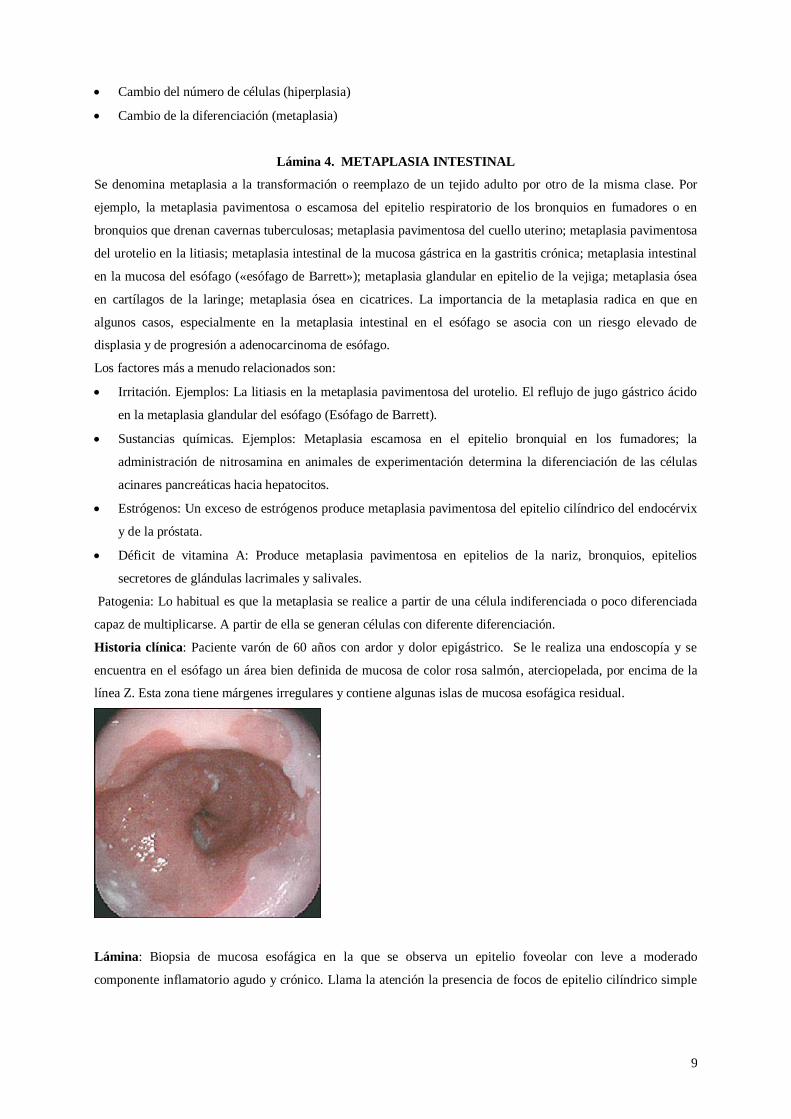
Historia clínica: Paciente varón de 60 años con ardor y dolor epigástrico. Se le realiza una endoscopía y se
encuentra en el esófago un área bien definida de mucosa de color rosa salmón, aterciopelada, por encima de la
línea Z. Esta zona tiene márgenes irregulares y contiene algunas islas de mucosa esofágica residual.
Lámina: Biopsia de mucosa esofágica en la que se observa un epitelio foveolar con leve a moderado
componente inflamatorio agudo y crónico. Llama la atención la presencia de focos de epitelio cilíndrico simple

10
con chapa estriada y células caliciformes, que corresponden a metaplasia intestinal, en continuidad con el
epitelio esofágico. No se observa cambios displásicos.
Lámina 5. HIPERPLASIA ENDOMETRIAL
Desde el punto de vista patogénico pueden distinguirse dos grandes grupos de hiperplasia: las secundarias a una
hipertrofia, y las primarias.
Las hiperplasias secundarias se explican por un desequilibrio trófico producido en la célula cuando la razón entre
volumen y superficie pasa un valor crítico. A medida que aumenta el radio de una célula, el volumen crece
proporcionalmente a la tercera potencia, mientras la superficie lo hace proporcionalmente a la segunda potencia
del radio.
Las hiperplasias primarias generalmente se deben a factores endocrinos, como la del endometrio, próstata y
glándula tiroides.
El sangrado uterino anormal debe hacer sospechar la posibilidad de un proceso proliferativo en la cavidad
endometrial, sobre todo si se acompaña de engrosamiento del endometrio (evidenciado por ecografía). En estos
casos está indicado realizar una biopsia para descartar la posibilidad de hiperplasia o neoplasia. Se estima que
gran parte de los cánceres de endometrio tienen como precursor una hiperplasia y que las hiperplasias
endometriales están relacionadas con producción incrementada de estrógenos. Las hiperplasias de endometrio se
clasifican como simples o complejas y se evalúa la presencia o ausencia de atipia en las glándulas endometriales
a fin de determinar la conducta clínica a seguir.
Historia clínica: Paciente mujer de 45 años con metrorragia (sangrado vaginal sin relación con ciclo menstrual).
En su ecografía se encuentra un endometrio engrosado.

11
Lámina: Biopsia de endometrio en la que se observa tejido endometrial con estructuras glandulares en fase
proliferativa, con un patrón anormal de proliferación caracterizado por presencia de mayor densidad de glándulas
endometriales, pérdida de la polaridad glandular (normalmente las glándulas proliferan longitudinalmente desde
la capa basal hacia la superficie del endometrio), dilatación glandular, sin atipia nuclear y con presencia de
mitosis. Los cambios en su conjunto corresponden a una hiperplasia endometrial simple sin atipia.
12
PRÁCTICA N° 2: PATOLOGIA CELULAR II
Lámina 6. HIPERTROFIA DE MÚSCULO LISO
Se habla de hipertrofia en sentido estricto cuando el aumento de masa protoplasmática induce aumento del
tamaño de la célula, concepto diferente de hiperplasia en la que hay aumento del número de células. Para poder
hablar de hiperplasia, tiene que tratarse de una estructura que directa o indirectamente pueda descomponerse en
células, es decir, tiene que tratarse al menos de un tejido. No puede hablarse de hiperplasia de una célula. El
concepto de hipertrofia en sentido amplio, al no hacer uso de ningún nivel de referencia, puede emplearse
indistintamente para órganos, tejidos o células aisladas. Dicho concepto es el que se emplea en la dimensión
macroscópica o, en general, cuando por cualquier razón no ha podido establecerse si hay o no aumento del
número de células, como por ejemplo, en la hipertrofia de la túnica media de las arterias.
El concepto de hipertrofia puede extenderse al nivel subcelular; así por ejemplo, puede hablarse de hipertrofia
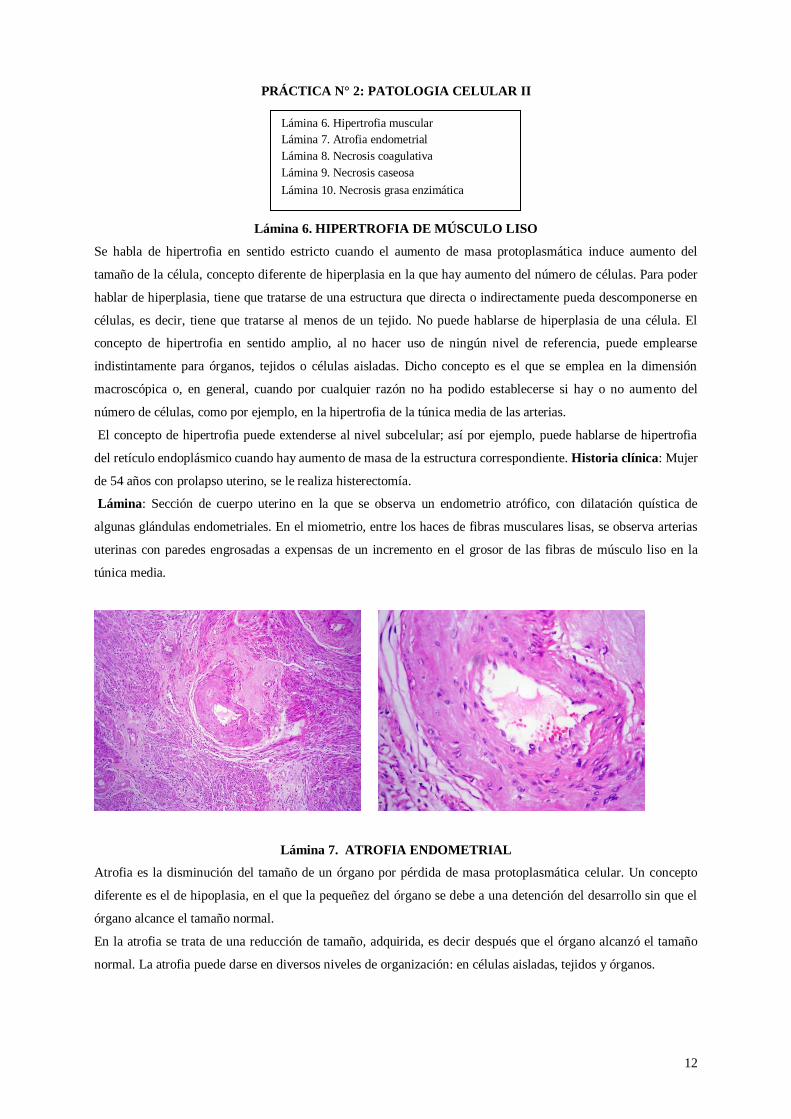
del retículo endoplásmico cuando hay aumento de masa de la estructura correspondiente. Historia clínica: Mujer
de 54 años con prolapso uterino, se le realiza histerectomía.
Lámina: Sección de cuerpo uterino en la que se observa un endometrio atrófico, con dilatación quística de
algunas glándulas endometriales. En el miometrio, entre los haces de fibras musculares lisas, se observa arterias
uterinas con paredes engrosadas a expensas de un incremento en el grosor de las fibras de músculo liso en la
túnica media.
Lámina 7. ATROFIA ENDOMETRIAL
Atrofia es la disminución del tamaño de un órgano por pérdida de masa protoplasmática celular. Un concepto
diferente es el de hipoplasia, en el que la pequeñez del órgano se debe a una detención del desarrollo sin que el
órgano alcance el tamaño normal.
En la atrofia se trata de una reducción de tamaño, adquirida, es decir después que el órgano alcanzó el tamaño
normal. La atrofia puede darse en diversos niveles de organización: en células aisladas, tejidos y órganos.
Lámina 6. Hipertrofia muscular
Lámina 7. Atrofia endometrial
Lámina 8. Necrosis coagulativa
Lámina 9. Necrosis caseosa
Lámina 10. Necrosis grasa enzimática
13
Un ejemplo de atrofia de órganos es la del cerebro; las superficies cortical y ependimaria tienden a acercarse
entre sí, los surcos se ensanchan y las circunvoluciones se adelgazan (especialmente en los lóbulos frontales), el
sistema ventricular se dilata y el espacio perivascular se amplía (perceptible a veces en forma de cribas).
La pérdida de masa protoplasmática referida al organismo entero se llama emaciación, marasmo o caquexia.
En la atrofia, la pérdida de masa protoplasmática afecta principalmente al parénquima de los órganos, por eso en
los órganos atróficos el estroma suele ser prominente y parecer aumentado. Como ocurre en el corazón atrófico,
en que los vasos, menos afectados que el miocardio, parecen demasiado grandes y las arterias coronarias se
hacen flexuosas. Esta desproporción entre el tamaño de los vasos y el del órgano puede servir para distinguir una
atrofia de una hipoplasia, como en el caso del riñón. La pérdida de masa protoplasmática en la atrofia se produce
lentamente a través de un proceso de desequilibrio entre anabolismo y catabolismo, en particular no se trata de
una pérdida de masa protoplasmática por necrosis, lo que representa en verdad una pseudoatrofia. Ejemplos de
pseudoatrofias son la llamada atrofia amarilla aguda del hígado y la atrofia roja subaguda del hígado, en que la
pérdida de masa protoplasmática se debe a una necrosis masiva; otro ejemplo es la llamada atrofia granular de la
corteza cerebral, en que se producen pequeñas depresiones debidas a necrosis selectiva de neuronas y
microinfartos.
No todas las atrofias son patológicas: existen ortoatrofias y patoatrofias. La ortoatrofia se ve en las gónadas,
útero y trompas después de la vida fértil y en general en diversos órganos dentro de los procesos involutivos de
la senectud, como en el bazo, en el timo, en las arterias y en diversos órganos. El corazón es uno de los pocos
órganos que no sufre atrofia senil. La ortoatrofia suele acompañarse de aumento de fibras colágenas, como en la
mucosa tubaria, en el miometrio y en los vasos. En las arterias disminuyen las fibras musculares y elásticas, lo
que se aprecia en la aorta como una disminución de la elasticidad.
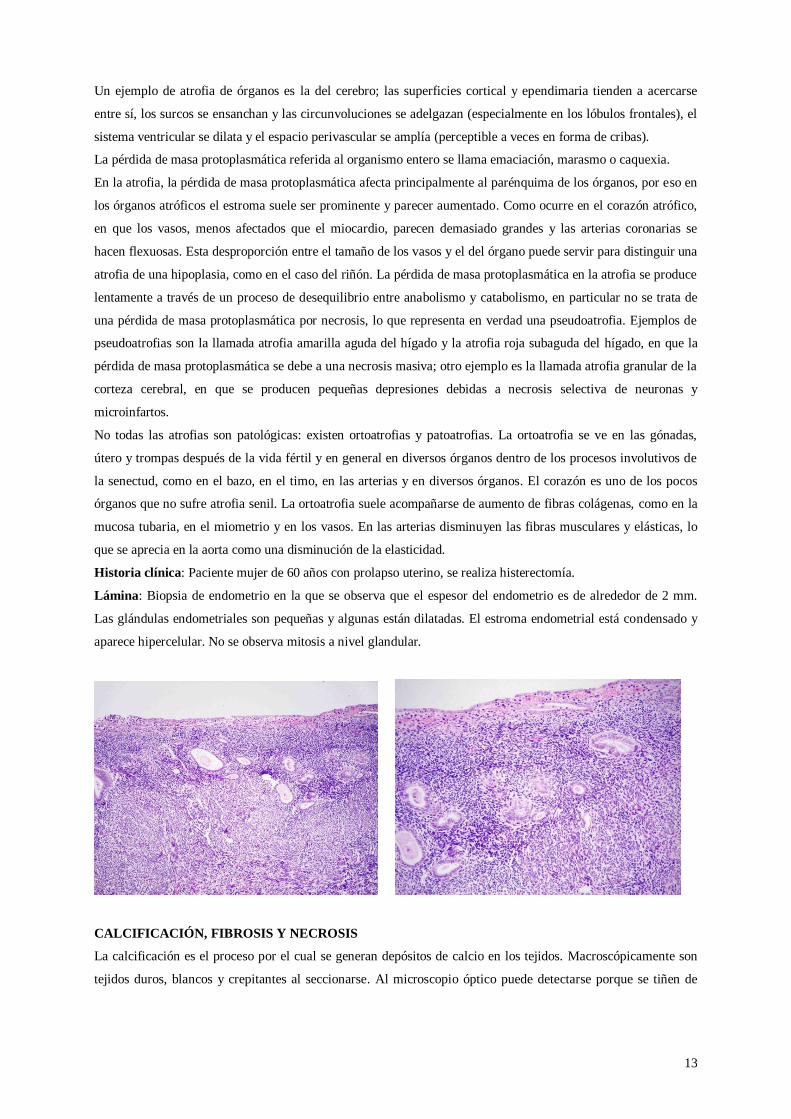
Historia clínica: Paciente mujer de 60 años con prolapso uterino, se realiza histerectomía.
Lámina: Biopsia de endometrio en la que se observa que el espesor del endometrio es de alrededor de 2 mm.
Las glándulas endometriales son pequeñas y algunas están dilatadas. El estroma endometrial está condensado y
aparece hipercelular. No se observa mitosis a nivel glandular.
CALCIFICACIÓN, FIBROSIS Y NECROSIS
La calcificación es el proceso por el cual se generan depósitos de calcio en los tejidos. Macroscópicamente son
tejidos duros, blancos y crepitantes al seccionarse. Al microscopio óptico puede detectarse porque se tiñen de

14
azul oscuro con hematoxilina/eosina. De forma fisiológica sólo están calcificados los huesos y los dientes; sin
embargo, pueden producirse calcificaciones fuera de estos tejidos, lo que recibe el nombre de calcificación
heterotópica. Existen dos tipos de calcificación heterotópica: la calcificación distrófica y la metastásica. La
calcificación distrófica se produce en tejidos anormales o degenerados al aumentar el calcio intracelular y se
suele observar en algunas arterias (calcificación de Monckeberg), en los cálculos (renales o vesiculares), cuando
mueren y se degeneran algunos microorganismos en los tejidos (como por ejemplo parásitos), en las cavernas de
la tuberculosis y en algunas neoplasias. La calcificación distrófica puede ser intra y/o extracelular y puede
progresar a la formación de tejido óseo (metaplasia ósea). El mecanismo de calcificación distrófica incluye dos
etapas: iniciación y propagación. Cuando una célula sufre necrosis, grandes cantidades de calcio ingresan a su
interior al fallar la bomba de sodio y potasio. Este calcio se combina con fosfatos dentro de las mitocondrias y se
producen cristales de hidroxiapatita. El mecanismo de las calcificaciones extracelulares es similar, pero en este
caso los cristales se forman en vesículas revestidas de membranas derivadas de células degeneradas. Después de
la iniciación, la propagación de la formación de cristales depende de la concentración local de calcio y fosfato.
La mayor parte de las veces la calcificación distrófica sólo es señal de lesión previa a los tejidos, aunque algunas
veces (como en el caso de las calcificaciones de las válvulas cardiacas) pueden afectar la función de estos.
La calcificación metastásica se produce en individuos con hipercalcemia sostenida. Esta hipercalcemia puede
estar asociada a hiperparatiroidismo, exceso de vitamina D o destrucción importante de tejido óseo por
metástasis óseas de tumores. Los órganos más afectados suelen ser el riñón, estómago, pulmones y vasos
sanguíneos.
Fibrosis. Es un término utilizado para referirse al resultado de la reparación celular, cuando las lesiones no están
activas y los mecanismos de cicatrización han dado como resultado la acumulación de fibras de colágeno para
reparar la lesión inicial. Desde el punto de vista histopatológico la fibrosis puede encontrarse en diversos
contextos, como por ejemplo en cicatrices hipertróficas, en secuelas de procesos inflamatorios crónicos de
órganos nobles como pielonefritis o glomerulonefritis o como componente principal del daño parenquimal en
entidades específicas como fibrosis pulmonar o cirrosis hepática.
Necrosis. La necrosis puede definirse como la muerte celular patológica reconocible por signos morfológicos.
Estos son: en el citoplasma, hipereosinofilia y pérdida de la estructura normal; en el núcleo, picnosis, cariolisis o
cariorrexis. La picnosis es la retracción del núcleo con condensación de la cromatina; la cariolisis, la disolución
del núcleo; la cariorrexis, la fragmentación del núcleo en trozos con cromatina condensada.
Las alteraciones del citoplasma y núcleo son coexistentes. La picnosis, cariolisis y cariorrexis no constituyen
etapas de la alteración nuclear; representan, aparentemente, formas distintas de reacción. En esta definición se
destacan dos ideas: por una parte, el carácter patológico de la necrosis como la manifestación más grave de
enfermedad a nivel celular; por otra, la base morfológica, dada por los signos para reconocimiento de la necrosis.
La primera idea excluye de la necrosis toda muerte celular que no sea manifestación de enfermedad, es decir la
apoptosis que ocurre en la muerte celular normal en los tejidos lábiles, es decir, en los que están sometidos
normalmente a un recambio de células, como los eritrocitos, las células epidérmicas, las células de los epitelios
respiratorio y digestivo, etcétera. Se excluye también la muerte celular dentro del proceso de remodelación de
órganos en desarrollo. No abarca tampoco la muerte celular que ocurre en el organismo muerto, como fenómeno
cadavérico. No comprende, por último, la muerte de células separadas del organismo y producida por la acción
de líquidos fijadores, pues dicha muerte no es manifestación de enfermedad.

15
La segunda idea excluye de la necrosis otras formas de muerte celular patológica o apoptosis asociada a
condiciones patológicas, por ejemplo, la muerte celular por la que puede producirse una atrofia numérica.
Lámina 8. NECROSIS COAGULATIVA
La principal diferencia histológica entre un tejido normal y un tejido con necrosis coagulativa es que el segundo
muestra pérdida de la tinción de los núcleos celulares y que sus citoplasmas se tiñen de un rosado más oscuro
con la eosina. Este es el patrón más común de necrosis, ocurre en muchos órganos sólidos y usualmente está
asociada a oclusión del riego arterial. . También se observa en neoplasias cuya velocidad de crecimiento excede
la capacidad de formar nuevos vasos sanguíneos para irrigar los tejidos neoplásicos, produciéndose necrosis
tumoral. La explicación de porqué la arquitectura básica y los límites celulares se preservan probablemente sea
porque la lesión destruye no solamente las proteínas estructurales vitales dentro de la membrana, citoplasma y
núcleo, sino también las enzimas dentro de los lisosomas que de otro modo degradarían los componentes
celulares y extracelulares. Los tejidos, de hecho, no permanecen en este estado por siempre; al poco tiempo
células inflamatorias son movilizadas al lugar de la lesión y estas digieren los componentes celulares, siendo los
detritus celulares removidos después por macrófagos.
Historia clínica: Paciente con una neoplasia maligna pulmonar. Se realiza una biopsia.
Lámina: Se observa una zona de transición entre el tejido con tumor viable (Carcinoma) y el tejido necrótico. En
la interfase se observa siluetas celulares correspondientes a células con necrosis coagulativa.

16
Lámina 9. NECROSIS CASEOSA
La necrosis caseosa (del latín caseum, queso) típicamente ocurre en tuberculosis y se llama así debido a la
apariencia de la sustancia necrótica, pastosa y de color blanco grisáceo que simula “queso suizo”. El análisis
químico revela otras semejanzas ya que tanto el queso como el material necrosado caseoso son ricos en lípidos y
proteínas. Se ha sugerido que la necrosis caseosa es característica de la necrosis tuberculosa por las siguientes
razones: 1) el bacilo de la tuberculosis contiene abundantes ceras y lípidos que quedan en los tejidos al morir
aquellos, 2) los ácidos grasos nacidos de la desintegración de estos lípidos inhiben las enzimas proteolíticas y se
conserva la proteína. El área necrótica no es líquida ni el contorno del tejido está preservado como en la necrosis
coagulativa. Al microscopio la sustancia tiene un aspecto amorfo debido a la desintegración completa del tejido;
sin embargo, en las primeras fases de su desarrollo pueden persistir durante cierto tiempo estructuras titulares
resistentes como fibras elásticas y colágenas.
Historia clínica: Paciente mujer de 53 años de edad con historia de tos, expectoración hemoptoica, baja de peso
y fiebre desde hace un año. Antecedente de cuadro similar hace cuatro años recibiendo tratamiento completo
para Tuberculosis pulmonar. Tiene adenopatías perihiliares que al corte dejan ver un material blanquecino con
necrosis. Se toma una biopsia.
Lámina: Biopsia ganglionar en la que se encuentran extensas áreas de necrosis de color rosado donde las células
muertas forman una masa proteínacea amorfa no pudiéndose apreciar arquitectura original. Las áreas necróticas
están rodeadas por un manguito linfohistiocítico constituyendo granulomas caseificantes, que son característicos
de la Tuberculosis. Además se encuentran áreas de fibrosis con presencia de infiltrado inflamatorio crónico.

17
Lámina 10. NECROSIS GRASA ENZIMÁTICA
Este tipo de necrosis es peculiar del tejido adiposo y es el tipo más frecuente de necrosis encontrado en la mama
después de un trauma y dentro de la grasa peritoneal como resultado de pancreatitis. En la mama un trauma
puede producir necrosis y ruptura de los adipocitos con liberación de su contenido lipídico que provoca una
reacción inflamatoria a cuerpo extraño, en virtud de la cual los macrófagos infiltran el territorio para fagocitar
los lípidos. Como los lípidos son poco solubles, la ligera reacción inflamatoria que se desarrolla puede durar
varias semanas, quedando toda el área tumefacta por lo que muchas veces se requiere excisión y examen
microscópico para determinar el diagnóstico. En la pancreatitis el daño de los acinos pancreáticos resulta en la
liberación de enzimas proteolíticas y lipolíticas que desnaturalizan las células adiposas en el peritoneo. Este
estado se presenta cuando las enzimas del páncreas experimentan activación antes de excretarse. Tal reacción
puede deberse a regurgitación de bilis por los conductos pancreáticos, especialmente si la bilis está infectada, a
traumatismos, infecciones o infartos del páncreas. Cualquier causa de muerte tisular, capaz de estimular la
actividad enzimática o que facilite el escape de la secreción pancreática desde los conductos a los espacios
intersticiales, puede desencadenar la digestión enzimática que una vez iniciada puede abarcar toda la glándula y
tejidos como epiplón y mesenterio. Cuando predominan los efectos de las enzimas proteolíticas se destruye gran
cantidad de tejido pancreático y esto se llama necrosis pancreática hemorrágica. Una reacción de esta magnitud
causa intensa peritonitis química, con grave desequilibrio hidroelectrolítico y shock, que suele terminar en la
muerte en uno o dos días. Más frecuentemente, el proceso se limita a una pequeña zona del páncreas y el
paciente se restablece. Al destruir la lipasa las células adiposas del tejido su contenido lipídico es liberado y se
acumula en los espacios intersticiales. Si prosigue la digestión enzimática la mayor parte de los lípidos son
desintegrados para formar ácidos grasos y glicerina. Los ácidos grasos reaccionan con el sodio, calcio y potasio,
para formar jabones que macroscópicamente aparecen como zonas blancas parecidas al yeso dentro del tejido
adiposo peri-pancreático. Como frecuentemente el jugo pancreático se derrama en la cavidad peritoneal, también
son afectados los tejidos adiposos del epiplón mayor y mesenterio donde suelen encontrarse placas blanquecinas.
En ocasiones puede encontrarse focos de necrosis grasa enzimática en sitios tan distantes como el mediastino y
el tejido celular subcutáneo. Esto se debe al transporte por los linfáticos de las enzimas pancreáticas. Algunas
enzimas como la amilasa y la lipasa pasan a la sangre y permiten valorarlas para diagnosticar pancreatitis.
Historia clínica: Paciente varón de 54 años con enfermedad actual de inicio agudo caracterizada por dolor
epigástrico, náuseas, vómitos, distensión abdominal, postración, fiebre y antecedente de alcoholismo crónico.
Dentro de los exámenes de laboratorio lo positivo es leucocitosis, amilasas y lipasas en suero y orina
aumentadas. El paciente es operado de urgencia y se encuentra extensa necrosis y hemorragia a nivel de cabeza
de páncreas. Se realiza una resección parcial.
18
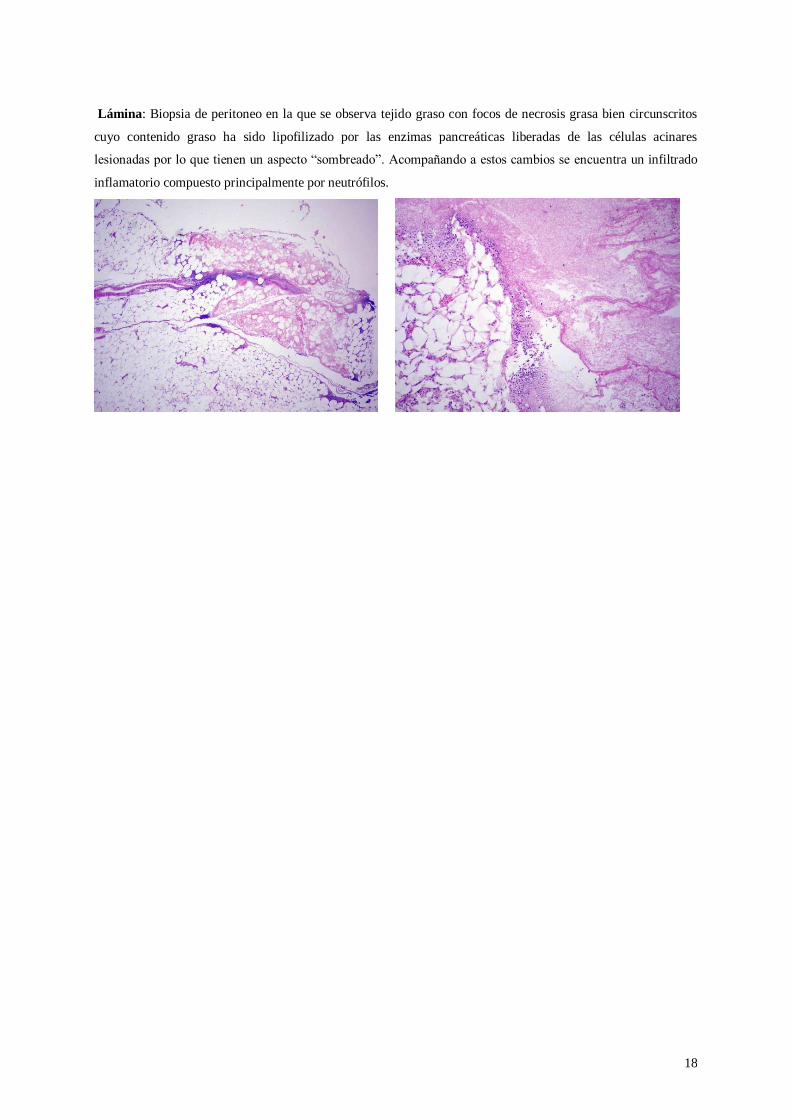
Lámina: Biopsia de peritoneo en la que se observa tejido graso con focos de necrosis grasa bien circunscritos
cuyo contenido graso ha sido lipofilizado por las enzimas pancreáticas liberadas de las células acinares
lesionadas por lo que tienen un aspecto “sombreado”. Acompañando a estos cambios se encuentra un infiltrado
inflamatorio compuesto principalmente por neutrófilos.

19
PRÁCTICA N° 3: INFLAMACIÓN AGUDA Y CRÓNICA
Cuando un tejido es lesionado, independientemente de su evolución final, la respuesta inicial es una reacción
inflamatoria aguda, siendo sus principales células efectoras los neutrófilos. Si el agente persiste y la lesión tisular
continúa los procesos de reparación y respuesta inflamatoria se dan en simultáneo como una reacción
inflamatoria crónica, cuyas principales células efectoras son los macrófagos.
La inflamación es un proceso con amplia variabilidad. El aspecto macroscópico fue caracterizado por Celso por
cuatro signos, que se conocen hoy como los signos cardinales de la inflamación: rubor, tumor, calor y dolor.
Posteriormente se agregó un quinto signo: la perturbación funcional.
La inflamación puede definirse como una reacción defensiva local integrada, con, exudación y proliferación. Se
le ha llamado «el síndrome local de adaptación». La reacción es desencadenada por estímulos nocivos de muy
diversa naturaleza: físicos, químicos y microorganismos como bacterias, hongos y parásitos. Una inflamación
puede conducir a la muerte del individuo si se desarrolla en órganos vitales. El calor y el rubor se explican por la
hiperemia activa que se produce en la inflamación; la tumoración, por el exudado; el dolor, por la irritación de
las terminaciones nerviosas producida por la alteración y el descenso del pH que acompaña al exudado.
Clínicamente se distinguen la inflamación aguda y la crónica. La inflamación aguda suele ser de iniciación
brusca, con síntomas muy manifiestos y de corta duración. La inflamación crónica suele ser de instalación
paulatina, de síntomas apagados y de larga duración. Las inflamaciones agudas son más frecuentemente
exudativas; las inflamaciones crónicas siempre tienen un componente productivo importante.
Existen, sin embargo, inflamaciones agudas predominantemente productivas linfoplasmocitarias, como algunas
formas de miocarditis y neumonitis intersticiales causadas por virus.
También hay inflamaciones crónicas con abundante exudado, como son las supuraciones crónicas, entre ellas las
osteomielitis crónicas purulentas y los abscesos. La salpingitis y la pielonefritis crónicas suelen tener abundante
exudado leucocitario.
A través de los efectos que se señalan a continuación puede entenderse el carácter defensivo local del proceso
inflamatorio. Ellos son:
Dilución de toxinas por el líquido del exudado;
Aporte de oxígeno y de anticuerpos por el flujo del exudado;
Remoción de metabolitos tóxicos por el flujo del exudado;
Formación de una red de fibrina, que delimita el proceso inflamatorio;
Estímulo del proceso inmunitario por transporte de microorganismos y toxinas a los ganglios linfáticos a
través del flujo del exudado, y
Fagocitosis, microfagia y macrofagia de agentes nocivos.
La fagocitosis se realiza en tres fases: adhesión, formación de la vacuola fagocítica y formación del fagosoma
(por fusión de un lisosoma con la vacuola fagocítica).
Lámina 11. Gastritis aguda y crónica
Lámina 12. Apendicitis
Lámina 13. Neumonía bacteriana
Lámina 14. Pielonefritis crónica
Lámina 15. Colecistitis crónica reagudizada

20
Lámina 11. INFLAMACIÓN: GASTRITIS AGUDA Y CRÓNICA
En la gastritis aguda suele observarse una constelación de cambios histológicos que dan una idea clara del
proceso patológico básico. Además de un infiltrado inflamatorio que suele contener neutrófilos, algunas veces
con formación de microabscesos glandulares, se puede observar otros cambios que reflejan la injuria sobre las
glándulas gástricas y que consisten en: depleción mucinosa, erosiones y cambios regenerativos a nivel de la capa
mucosa, principalmente. Normalmente a todo lo largo de la mucosa del aparato digestivo se puede observar
algunos linfocitos y células plasmáticas que cumplen roles de vigilancia inmunológica; sin embargo, la presencia
de acúmulos linfoides primarios o secundarios en la mucosa digestiva (con excepción de las Placas de Peyer de
la región ileal) siempre se considera como un marcador de proceso inflamatorio crónico. Los principales
cambios relacionados con gastritis son la presencia de Helicobacter pylori colonizando la superficie mucosa.
Cambios tardíos que reflejan una injuria sostenida son la atrofia glandular y la presencia de metaplasia intestinal.
Es bueno tener presente que en la patogenia del adenocarcinoma gástrico de tipo intestinal, la metaplasia
intestinal y la atrofia glandular son factores que se relacionan muy bien con la ocurrencia de gastritis aguda y
crónica y sus complicaciones.
Historia Clínica. Paciente varón de 56 años con dolor abdominal tipo ardor que inicialmente se presentaba
cuando no almorzaba a su hora acostumbrada y que cedía con la ingesta de alimentos. En las últimas semanas
este dolor estaba presente por las mañanas al despertarse y cedía temporalmente con el uso de antiácidos. Se le
realiza una endoscopía digestiva y se observa eritema difuso a nivel de la región antropilórica. Se toma biopsia.
Lámina. Se observa una mucosa gástrica con edema y congestión vascular. La capa de moco superficial aparece
parcialmente depletada; además se observa un moderado infiltrado inflamatorio en la lámina propia constituido
por neutrófilos y linfocitos. Hay acúmulos linfoides primarios y leve atrofia glandular. Se observa Helicobacter
pylori.

21
Lámina 12. INFLAMACIÓN AGUDA: APENDICITIS AGUDA
La apendicitis aguda es una de las principales causas de intervenciones quirúrgicas de emergencia. Se presenta
sin relación aparente con un factor desencadenante y es más frecuente en pacientes jóvenes, aunque también se
puede presentar en niños, adultos y ancianos. Los pacientes usualmente se quejan de dolor abdominal intenso,
usualmente referidos al cuadrante inferior derecho del abdomen el que, al progresar se acompaña de signos de
irritación peritoneal, el cual puede ser demostrado clínicamente. Adicionalmente, los pacientes suelen tener
alteraciones en el hemograma, el cual suele mostrar leucocitosis con desviación izquierda (mayor número de
formas juveniles).
Historia clínica: Paciente varón de 17 años con una enfermedad de 18 horas caracterizada por dolor abdominal
de inicio epigástrico que luego se localiza en fosa ilíaca derecha acompañado de náuseas, vómitos y fiebre. En el
examen abdominal se encuentran ruidos hidroaéreos disminuidos y signo de Mac Burney. En el hemograma se
encontró leucocitosis y desviación izquierda (abastonados 8%). Se realiza una laparotomía y apendicectomía. El
apéndice estaba congestivo, parcialmente cubierto por una capa de fibrina amarillenta y con áreas necróticas a
predominio distal.
Lámina: Sección de apéndice cecal. Adherido a la serosa se encuentra una membrana fibrinoleucocitaria. Se
observa un infiltrado inflamatorio que compromete la mucosa, la capa muscular y la serosa. A mayor aumento se
puede observar células inflamatorias, predominantemente polimorfonucleares, en proceso de adhesión y
migración a través de las paredes endoteliales.
22
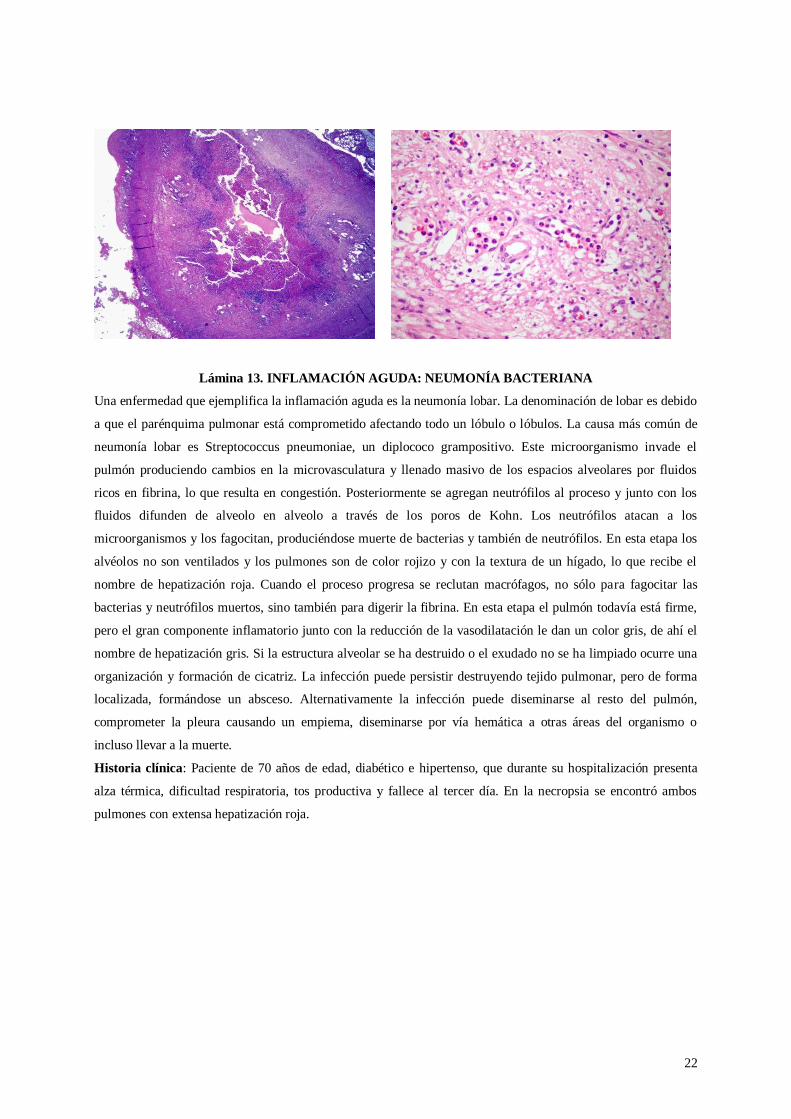
Lámina 13. INFLAMACIÓN AGUDA: NEUMONÍA BACTERIANA
Una enfermedad que ejemplifica la inflamación aguda es la neumonía lobar. La denominación de lobar es debido
a que el parénquima pulmonar está comprometido afectando todo un lóbulo o lóbulos. La causa más común de
neumonía lobar es Streptococcus pneumoniae, un diplococo grampositivo. Este microorganismo invade el
pulmón produciendo cambios en la microvasculatura y llenado masivo de los espacios alveolares por fluidos
ricos en fibrina, lo que resulta en congestión. Posteriormente se agregan neutrófilos al proceso y junto con los
fluidos difunden de alveolo en alveolo a través de los poros de Kohn. Los neutrófilos atacan a los
microorganismos y los fagocitan, produciéndose muerte de bacterias y también de neutrófilos. En esta etapa los
alvéolos no son ventilados y los pulmones son de color rojizo y con la textura de un hígado, lo que recibe el
nombre de hepatización roja. Cuando el proceso progresa se reclutan macrófagos, no sólo para fagocitar las
bacterias y neutrófilos muertos, sino también para digerir la fibrina. En esta etapa el pulmón todavía está firme,
pero el gran componente inflamatorio junto con la reducción de la vasodilatación le dan un color gris, de ahí el
nombre de hepatización gris. Si la estructura alveolar se ha destruido o el exudado no se ha limpiado ocurre una
organización y formación de cicatriz. La infección puede persistir destruyendo tejido pulmonar, pero de forma
localizada, formándose un absceso. Alternativamente la infección puede diseminarse al resto del pulmón,
comprometer la pleura causando un empiema, diseminarse por vía hemática a otras áreas del organismo o
incluso llevar a la muerte.
Historia clínica: Paciente de 70 años de edad, diabético e hipertenso, que durante su hospitalización presenta
alza térmica, dificultad respiratoria, tos productiva y fallece al tercer día. En la necropsia se encontró ambos
pulmones con extensa hepatización roja.
23
Lámina: A menor aumento se observa alveolos llenos de células inflamatorias, los septos muestran edema y
congestión vascular. A mayor aumento los alvéolos pulmonares se encuentran llenos de polimorfonucleares y
macrófagos. En algunos vasos sanguíneos se puede observar el fenómeno de adherencia de los leucocitos al
endotelio.
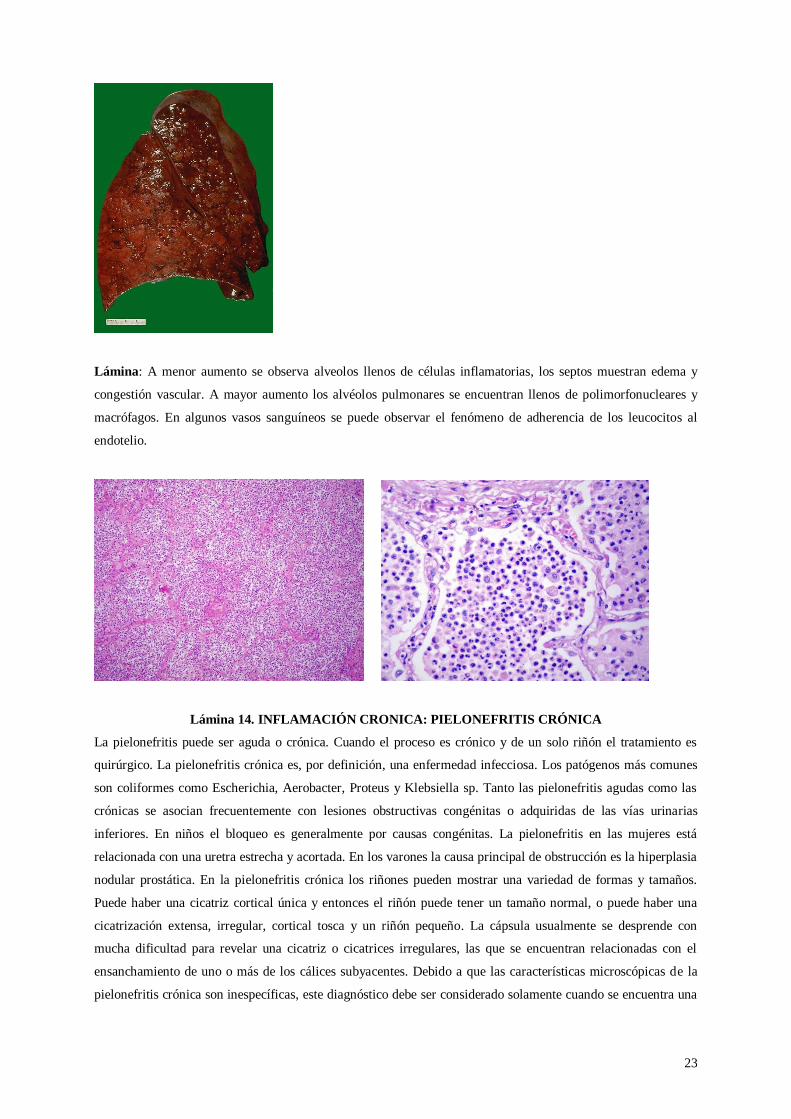
Lámina 14. INFLAMACIÓN CRONICA: PIELONEFRITIS CRÓNICA
La pielonefritis puede ser aguda o crónica. Cuando el proceso es crónico y de un solo riñón el tratamiento es
quirúrgico. La pielonefritis crónica es, por definición, una enfermedad infecciosa. Los patógenos más comunes
son coliformes como Escherichia, Aerobacter, Proteus y Klebsiella sp. Tanto las pielonefritis agudas como las
crónicas se asocian frecuentemente con lesiones obstructivas congénitas o adquiridas de las vías urinarias
inferiores. En niños el bloqueo es generalmente por causas congénitas. La pielonefritis en las mujeres está
relacionada con una uretra estrecha y acortada. En los varones la causa principal de obstrucción es la hiperplasia
nodular prostática. En la pielonefritis crónica los riñones pueden mostrar una variedad de formas y tamaños.
Puede haber una cicatriz cortical única y entonces el riñón puede tener un tamaño normal, o puede haber una
cicatrización extensa, irregular, cortical tosca y un riñón pequeño. La cápsula usualmente se desprende con
mucha dificultad para revelar una cicatriz o cicatrices irregulares, las que se encuentran relacionadas con el
ensanchamiento de uno o más de los cálices subyacentes. Debido a que las características microscópicas de la
pielonefritis crónica son inespecíficas, este diagnóstico debe ser considerado solamente cuando se encuentra una

24
cicatriz cortical con una deformidad pielocalicial subyacente. Esta deformidad también es reconocible por el
radiólogo. La característica microscópica más llamativa de la pielonefritis crónica es la distribución en placa de
las lesiones. En las áreas afectadas los cambios están presentes en los glomérulos, túbulos, intersticio y vasos.
Los glomérulos pueden mostrar un engrosamiento de la cápsula de Bowman y aún una fibrosis concéntrica por
fuera de la cápsula (fibrosis periglomerular). El ovillo glomerular puede estar colapsado, pero más comúnmente
muestra una esclerosis segmentaria asociada con un incremento de las células mesangiales, adherencias
capsulares y hialinosis segmentaria. Puede haber cambios secundarios a hipertensión. Los túbulos están
destruidos o atróficos. Muchos están dilatados y contienen cilindros eosinofílicos homogéneos. El intersticio está
infiltrado por un número variable de linfocitos, células plasmáticas y otras células inflamatorias. Siempre hay
fibrosis. En la mayoría de los casos las arterias muestran engrosamiento medial e intimal. La pelvis con
frecuencia contiene folículos linfoides, pero puede ser normal en los casos de nefropatía por reflujo.
Historia clínica. Paciente mujer con episodios de infecciones recurrentes de vías urinarias altas y bacteriuria
persistente, resistente a tratamiento antibiótico. La ecografía renal reveló un riñón derecho de 15 cm. de longitud
y en la urografía excretora se encontró defectos en la perfusión y aclaramiento renal. Se realizó nefrectomía
derecha. Se muestra la sección transversal de un riñón con una superficie deformada por lesiones cicatrizales.
Lámina. Se observa un parénquima renal con arquitectura básicamente conservada, llamando la atención la
presencia a menor aumento de acúmulos celulares en el intersticio renal. A mayor aumento se observa que esos
acúmulos celulares corresponden a células mononucleadas (linfocitos y macrófagos) y algunos
polimorfonucleares. Es posible observar cambios secundarios en el sistema tubular, los que se encuentran algo
atróficos y con cilindros granulosos y hialinos en sus luces.

25
LÁMINA Nº 15. INFLAMACIÓN CRÓNICA. COLECISTITIS CRÓNICA
La colecistitis crónica es una enfermedad muy frecuente y suele estar relacionada con la presencia de cálculos en
la vesícula biliar (colelitiasis). El marcador histológico de la inflamación crónica es la presencia de alteración
estructural en los tejidos, consecuencia de la destrucción y reparación tisular. En el caso de la colecistitis crónica
la evidencia más objetiva de la injuria crónica de la vesícula biliar lo constituye el engrosamiento de su pared,
que puede engrosarse notablemente como consecuencia de episodios de obstrucción al flujo de salida de la bilis
ocasionados por los cálculos biliares o también por el hecho de que al ocupar espacio dentro de la vesícula este
órgano tenga que realizar mayor esfuerzo para expulsar la bilis.
Historia Clínica. Paciente mujer de 38 años con historia de cólicos biliares esporádicos que en los últimos
meses aumentan de frecuencia e intensidad. En su ecografía se observa numerosos cálculos dentro de su vesícula
biliar. Es programada para una colecistectomía laparoscópica en la que se le extrae una vesícula de 8 cm. de
longitud conteniendo numerosos cálculos pardo-verdosos facetados en promedio de 1 cm. de longitud mayor. La
pared de la vesícula biliar medía 1 cm. de espesor y era de consistencia aumentada.
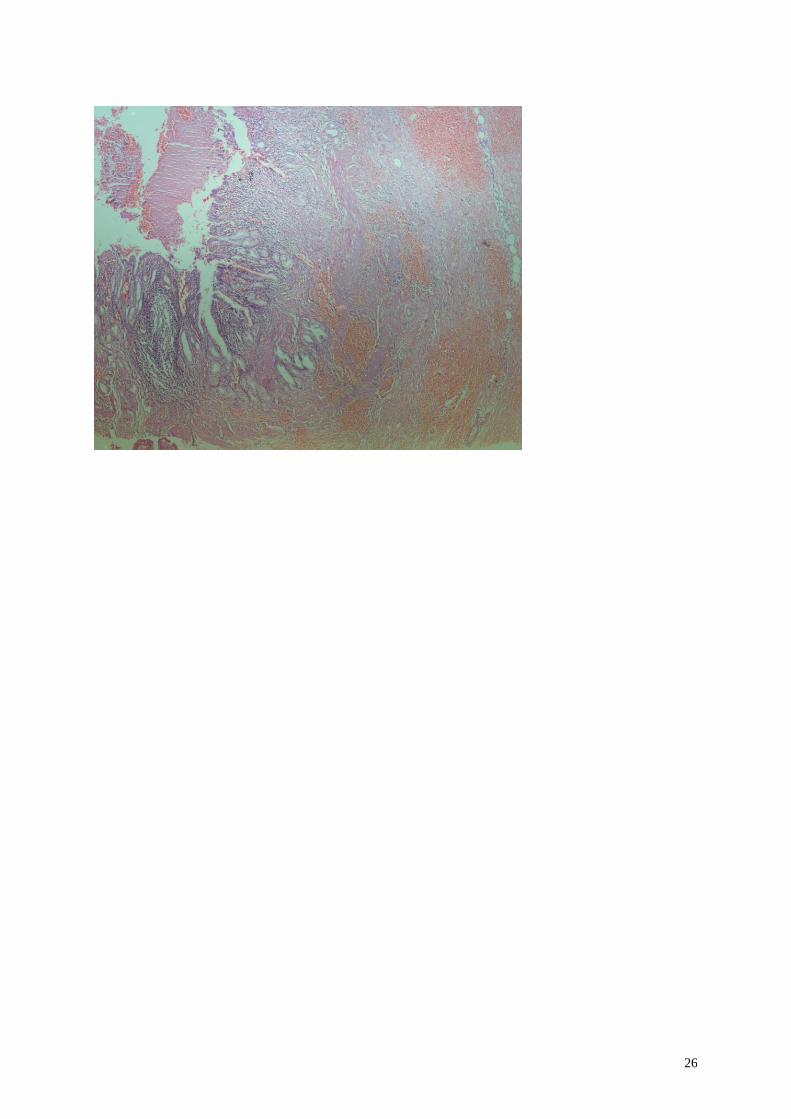
Lámina. Se observa una vesícula biliar de paredes engrosadas, de hasta 1 cm. de espesor, con criptas glandulares
que penetran en el espesor de la pared de músculo liso (senos de Rokitansky), hay ulceración focal de la mucosa,
áreas de hemorragia y focos dispersos de infiltrado inflamatorio crónico con formación de acúmulos linfoides
primarios. El engrosamiento de la pared vesicular se debe tanto a hipertrofia de la musculatura lisa como a la
presencia de tractos fibrosos de tipo cicatrizal que se extienden y confluyen en medio de las fibras musculares.
26

27
PRÁCTICA Nº 4. TRANSTORNOS HEMODINÁMICOS
Lámina 16. INFARTO PULMONAR: HEMORRAGIA
Infarto es la necrosis del parénquima y estroma producida por isquemia. La necrosis del estroma se acompaña de
destrucción del material intercelular. La palabra infarto viene del latín infarcire, que significa rellenar (infarctum
= rellenado). En efecto, en un infarto reciente la zona comprometida aparece tumefacta como rellena de fibrina.
El infarto es por lo tanto una lesión que se da sólo a nivel de órgano o de estructuras organoides, como una
mucosa. No pueden infartarse células. El concepto, además, lleva implícita una determinada patogenia: la
isquemia.
Forma del infarto: El infarto adopta aproximadamente la forma del territorio vascular comprometido en el
espesor del órgano. Puesto que a partir de una arteria las ramificaciones se suceden abarcando un espacio cada
vez mayor, el infarto tiene una forma parecida al de una pirámide o una cuña. Se le describe típicamente como
cuneiforme. El vértice está hacia la zona arterial obstruida, la base corresponde al territorio distal irrigado. La
obstrucción suele estar antes del vértice.
Cuando en el espesor de un órgano las ramificaciones arteriales son cortas y se disponen más o menos en la
misma dirección, la forma del infarto es laminar, como en la corteza cerebral o en el intestino. En el riñón el
infarto por obstrucción de una arteria arciforme (porque sus ramas nacen casi en ángulo recto) tiene la forma de
una pirámide trunca; en cambio los producidos por obstrucción de una arteria lobular o interlobulillar son cuñas
completas.
Disposición del infarto: La disposición del infarto depende del árbol vascular. En los órganos que tienen hilio,
las arterias atraviesan el espesor del órgano desde el hilio a la periferia a medida que se van ramificando. El
territorio terminal se halla en la superficie externa, donde se producirá la base de la cuña infartada. Este modelo
vale para el infarto renal, el esplénico y el pulmonar. La posición del vértice depende del calibre de la arteria
obstruida, suele estar en el espesor del órgano, pero en los grandes infartos se encuentra cerca del hilio. Otros
órganos carecen de hilio y las arterias se distribuyen por la superficie externa y desde allí penetran en el espesor.
Este modelo se da en los órganos huecos, como el corazón y el intestino. En el corazón la base de la cuña
infartada se encuentra hacia el endocardio; el vértice, hacia el epicardio. En el intestino el territorio terminal
corresponde a la mucosa. En el encéfalo y la médula espinal el sistema de irrigación es similar al del corazón.
Causas de isquemia en el infarto: La causa más frecuente es la obstrucción arterial por embolía trombótica o por
trombosis. La embolia trombótica es la causa más frecuente de los infartos pulmonares, renales y esplénicos. La
inmensa mayoría de los infartos del miocardio se debe a trombosis. En los infartos cerebrales la embolia
trombótica es más frecuente que la trombosis.
Existen infartos sin oclusión arterial (obstrucción arterial completa). Los factores más importantes que
condicionan estos infartos son la estenosis arterial y la hipotensión. Otro factor importante en el corazón es una
brusca demanda de trabajo que exija un aumento de flujo mayor que la reserva coronaria. En estos infartos se
Lámina 16. Infarto pulmonar
Lámina 17. Trombosis de la orejuela
Lámina 18. Edema pulmonar
Lámina 19. Infarto de miocardio
Lámina 20. Congestión vascular esplénica

28
compromete el sector que tiene mayor riesgo de que la irrigación se haga insuficiente. Estos sectores son los
territorios terminales y los limítrofes; territorios terminales son, en el intestino la mucosa; en el miocardio las
capas subendocárdicas. Es probable que en algunos territorios limítrofes, como en los cerebrales superficiales,
puedan producirse infartos por insuficiencia circulatoria periférica, como en el shock, sin que medie otro factor,
aparte del dado por la disposición particular del árbol arterial y la hipotensión.
Los tipos fundamentales de infarto son el anémico y el hemorrágico. Además, pueden distinguirse el infarto
séptico y el llamado infarto venoso.
Infarto anémico: Los infartos anémicos se producen en órganos con irrigación de tipo arboriforme (terminal). A
ellos pertenece el corazón, el riñón y el bazo. El infarto en estos órganos presenta sin embargo un aspecto
hemorrágico en las primeras 24 horas. Ello se debe a la extravasación de sangre contenida en los vasos que se
necrosan. A partir del segundo día, a medida que se reabsorbe esta sangre, van apareciendo los caracteres
macroscópicos típicos de la necrosis de coagulación. Se trata por lo tanto de un componente hemorrágico inicial
y sólo pasajero, que no quita el carácter de anémico de estos infartos, puesto que a la zona comprometida no
sigue llegando sangre. En la substancia blanca cerebral el infarto anémico no suele ser hemorrágico ni siquiera al
comienzo debido a que los vasos son poco numerosos.
Infarto hemorrágico: El hecho general que explica el carácter hemorrágico de algunos infartos es que sigue
llegando sangre a la zona isquémica aunque en cantidad insuficiente para mantener la vitalidad de los tejidos.
Las condiciones más frecuentes en que se da este hecho son:
obstrucción parcial,
migración de un émbolo trombótico,
irrigación de tipo anastomótico (reticular) y
doble circulación
Infarto séptico: Se produce por una embolia trombótica infectada. La lesión está constituida en parte por un
infarto, en parte por una inflamación con frecuencia purulenta. Los infartos sépticos suelen ser múltiples y
pequeños por la tendencia del émbolo a disgregarse en pequeñas masas debido a los gérmenes. El infarto séptico
pulmonar es menos hemorrágico que el infarto puro y no tiene como condición una hiperemia pasiva.
Infarto venoso: Se denomina así la infiltración hemorrágica de un órgano o de un sector de él, producida por el
bloqueo brusco del drenaje venoso. El tejido comprometido se disgrega, las células muestran alteraciones
tróficas y luego necrosis. Las causas más frecuentes son la trombosis venosa, como en el riñón y encéfalo, y la
torsión del pedículo vascular, como en el testículo y anexos uterinos. La torsión afecta más a las venas que
arterias. La denominación es impropia puesto que la lesión no se produce primariamente por isquemia sino por
hiperemia venosa.
El tejido necrótico del infarto desencadena a su alrededor primero una reacción inflamatoria, que a partir de las
24 horas se manifiesta en una infiltración leucocitaria. La liberación de enzimas leucocitarias contribuye a la lisis
del tejido necrótico. En la segunda mitad de la primera semana se produce una proliferación de macrófagos que
inician la remoción de los detritus. Aproximadamente después de una semana aparece tejido de granulación
alrededor del infarto; a medida que avanza el proceso de reabsorción y reparación se va retrayendo la zona
infartada que termina con el reemplazo por una cicatriz.
En los infartos con necrosis licuefactiva, como en el encéfalo, queda una cavidad con escaso material líquido.

29
Lámina 16. INFARTO PULMONAR
Historia clínica: Paciente de 75 años, que se encuentra postrado en cama desde hace un año por presentar
fractura de fémur, desarrolla un cuadro de insuficiencia respiratoria aguda con dolor torácico intenso y fallece al
tercer día debido a tromboembolia pulmonar. En la necropsia se encuentra un área extensa de color blanquecino
(infarto blanco) y un foco hemorrágico a nivel de la base (infarto rojo) del pulmón derecho y un trombo a nivel
de la arteria pulmonar derecha.
Lámina: Se observa la interfase entre tejido pulmonar preservado y la extensa zona con infarto hemorrágico. Al
examen microscópico se observa focos de hemorragia y edema alveolar (material eosinófilo en la luz alveolar).
Es notoria la presencia de un componente inflamatorio agudo (neumonía) en la vecindad del área infartada,
aunque no es posible definir desde el punto de vista histológico si es que la neumonía está relacionada con el
origen del infarto hemorrágico (infarto séptico) o es un fenómeno que se sobreagregó al cuadro clínico inicial.
Lámina 17. TROMBOSIS DE OREJUELA AURICULAR
Trombosis es la formación de una masa hemática sólida dentro de los vasos y durante la vida. La masa se llama
trombo. No hay que confundir la trombosis con la coagulación de la sangre, ni el trombo con los coágulos.
Durante la vida, la coagulación ocurre fuera de los vasos, ya sea dentro del organismo, como en los hematomas,
ya sea fuera de él, como en el tubo de ensayo. En ambos casos el mecanismo de la coagulación se desencadena
por liberación de tromboplastina principalmente de las plaquetas a medida que se destruyen al adherirse a los
tejidos y paredes del tubo. Después de la muerte se producen coágulos dentro de los vasos por liberación de

30
tromboplastina de las células endoteliales a medida que se desprenden de los vasos y lisan. Macroscópicamente
hay dos tipos: el trombo rojo o de coagulación y el trombo blanco o de aposición. Además, se distinguen los
microtrombos, que se producen en la microcirculación, especialmente en vénulas y capilares. En todos los
trombos el componente principal es la fibrina, pero desde el punto de vista de la patogenia, las plaquetas
desempeñan el papel primordial. En el trombo rojo los elementos figurados de la sangre, incluyendo la fibrina, se
encuentran mezclados desordenadamente. El trombo blanco típico es de aspecto coraliforme dado por finas
crestas transversales (capas plaquetarias que alternan con capas de leucocitos) que alternan con zonas
deprimidas. Las zonas deprimidas, más anchas que las salientes, están formadas por una red de fibrina que
contiene eritrocitos. Los factores patogenéticos principales son tres: lentitud de la corriente sanguínea, lesión
endotelial y alteraciones hematológicas que afectan la coagulación. Los microtrombos son manifestación de
alteraciones generales de la coagulación. Los posibles procesos que pueden ocurrir en un trombo son:
organización con recanalización del vaso, calcificación (distrófica), fragmentación y migración, infección, por
último, crecimiento por formación de nuevas masas trombóticas.
Complicaciones: Las principales son tres: obstrucción, embolía trombótica e infección.
Historia clínica. Paciente de 60 años de edad, hipertenso con diagnóstico de insuficiencia cardiaca avanzada
asociada a fibrilación auricular. El paciente fallece y en la necropsia se encuentra un gran trombo organizado a
nivel de la orejuela.
Lámina: Sección histológica de orejuela. Se observa tejido muscular cardiaco, endocardio (señalado con flecha
roja) con destrucción parcial y adherencias de trombo (flecha verde) constituido por material hialino con
hematíes y células degeneradas. En la zona adyacente al endocardio se observa organización del trombo
(reemplazo de la fibrina y hematíes por áreas hialinizadas con depósitos de material colágeno), en esa misma
zona hay recanalización del trombo (flecha azul) que consiste en la formación de neovasos en medio de los
depósitos de colágeno, lo que indica la antigüedad de la lesión.

31
Lámina 18. EDEMA PULMONAR
El término edema designa la acumulación de cantidades anormales de fluidos en los espacios intercelulares o en
cavidades corporales. Puede ser parte de un proceso localizado o generalizado. Cuando el edema es severo y
generalizado produce una marcada hinchazón de los tejidos subcutáneos y es llamado anasarca. Las colecciones
edematosas en las cavidades serosas reciben designaciones especiales como hidrotórax (pleura), hidropericardio
(pericardio) o ascitis (peritoneo). El edema no inflamatorio, que usualmente se origina de transtornos
hidrodinámicos es un trasudado, con bajo contenido de proteínas y otros coloides. Por el contrario, el edema
inflamatorio es rico en proteínas. Al nivel más elemental, el edema resulta de cualquier aumento en las fuerzas
que movilizan fluidos del compartimiento intravascular hacia el intersticial. Estas fuerzas son principalmente las
presiones hidrostáticas dentro de los vasos ayudadas en menor medida por la presión osmótica de los fluidos
fuera de los vasos sanguíneos. Las fuerzas que tienden a mantener los fluidos dentro de los compartimentos
vasculares son principalmente la presión osmótica de los coloides y, en menor medida, las presiones titulares
alrededor de los vasos sanguíneos. El edema pulmonar generalmente está asociado a insuficiencia cardiaca por
falla ventricular izquierda que produce progresiva acumulación de sangre en la circulación pulmonar.
Normalmente la presión hidrostática en los capilares pulmonares se encuentra en un rango entre 6 a 9 mmHg.
Cuando esta presión aumenta hasta 25 a 30 mm Hg ocurre congestión vascular seguida de edema franco. El
pulmón es particularmente vulnerable al desarrollo de edema debido a que su estructura en panal de abejas no
ejerce presión tisular significativa contra el escape de fluidos. Primero el trasudado está limitado alrededor de los
vasos, posteriormente se produce engrosamiento de las paredes vasculares a medida que el fluido se acumula en
ellas y finalmente el trasudado inunda los alveolos. En ocasiones el trasudado se acumula en el espacio pleural
produciendo derrame pleural.
Historia Clínica. Varón de 65 años con hipertensión arterial no controlada que llega al servicio de emergencia
con dificultad respiratoria severa en reposo, tos intensa y expectoración espumosa rosada. Se le toma una
radiografía simple de tórax en la que se halla densidades lineales y nodulares finas y cardiomegalia. Un
electrocardiograma es compatible con hipertrofia ventricular derecha. El paciente fallece al poco tiempo y en la
necropsia se encuentra líquido espumoso en tráquea y bronquios, los pulmones se hallan pesados, y a la sección
resuma líquido levemente sanguinolento. Los pulmones semejan esponjas llenas de líquido.
32
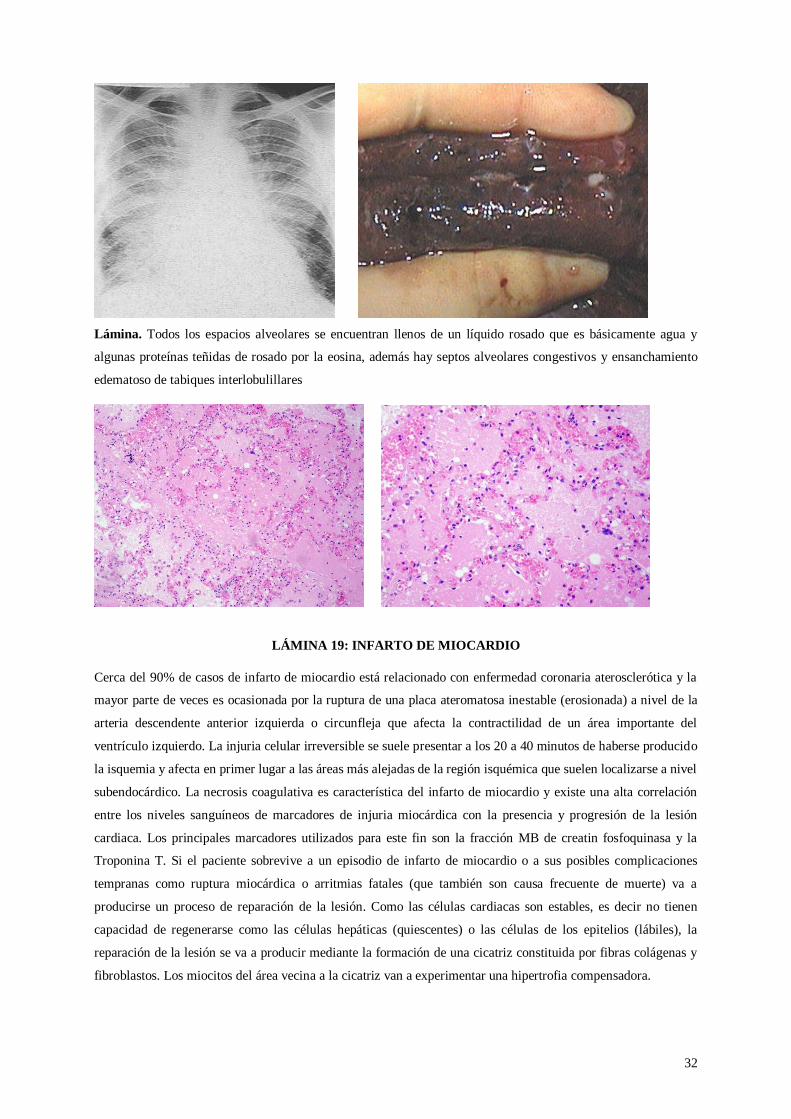
Lámina. Todos los espacios alveolares se encuentran llenos de un líquido rosado que es básicamente agua y
algunas proteínas teñidas de rosado por la eosina, además hay septos alveolares congestivos y ensanchamiento
edematoso de tabiques interlobulillares
LÁMINA 19: INFARTO DE MIOCARDIO
Cerca del 90% de casos de infarto de miocardio está relacionado con enfermedad coronaria aterosclerótica y la
mayor parte de veces es ocasionada por la ruptura de una placa ateromatosa inestable (erosionada) a nivel de la
arteria descendente anterior izquierda o circunfleja que afecta la contractilidad de un área importante del
ventrículo izquierdo. La injuria celular irreversible se suele presentar a los 20 a 40 minutos de haberse producido
la isquemia y afecta en primer lugar a las áreas más alejadas de la región isquémica que suelen localizarse a nivel
subendocárdico. La necrosis coagulativa es característica del infarto de miocardio y existe una alta correlación
entre los niveles sanguíneos de marcadores de injuria miocárdica con la presencia y progresión de la lesión
cardiaca. Los principales marcadores utilizados para este fin son la fracción MB de creatin fosfoquinasa y la
Troponina T. Si el paciente sobrevive a un episodio de infarto de miocardio o a sus posibles complicaciones
tempranas como ruptura miocárdica o arritmias fatales (que también son causa frecuente de muerte) va a
producirse un proceso de reparación de la lesión. Como las células cardiacas son estables, es decir no tienen
capacidad de regenerarse como las células hepáticas (quiescentes) o las células de los epitelios (lábiles), la
reparación de la lesión se va a producir mediante la formación de una cicatriz constituida por fibras colágenas y
fibroblastos. Los miocitos del área vecina a la cicatriz van a experimentar una hipertrofia compensadora.

33
Historia Clínica. Paciente varón de 56 años, hipertenso, con antecedente de infarto de miocardio 5 años atrás.
Fallece por edema agudo de pulmón debido a insuficiencia cardiaca congestiva descompensada. En los cortes
transversales del corazón se observa una pared miocárdica engrosada con áreas blanquecinas fibróticas a nivel
del ventrículo izquierdo.
Lámina. Se observa áreas hipocelulares, de aspecto cicatrizal, constituidas por fibroblastos y fibras colágenas en
medio de fibras musculares cardiacas hipertróficas y con núcleos hipercromáticos. Los hallazgos corresponden a
un infarto antiguo de miocardio. Los cambios cicatrizales son posteriores a la necrosis coagulativa que
caracteriza al infarto de miocardio y los cambios observados en las fibras cardiacas adyacentes corresponden a
mecanismos compensadores para suplir la inactividad de las fibras dañadas.
LÁMINA 20: CONGESTIÓN VASCULAR ESPLÉNICA
El bazo es un órgano hematolinfoide. Su función inmune es desempeñada por la pulpa blanca que aparece a
modo de puntos dispersos por todo el órgano, estos están constituidos por acúmulos linfoides primarios y
secundarios en cuyo centro aparece una rama de la arteria esplénica. La pulpa roja ocupa mayor espacio dentro
del órgano y tiene funciones relacionadas con la destrucción de células sanguíneas dañadas o envejecidas y con
el almacenamiento de sangre. En la pulpa roja se observa los llamados senos medulares y cordones de Billroth.
La esplenomegalia se refiere al aumento del tamaño del bazo y puede llegar a ser de gran magnitud. Las causas
más frecuentes de esplenomegalia están en relación con congestión pasiva esplénica, usualmente relacionada con
insuficiencia cardiaca congestiva y cirrosis hepática, y con enfermedades autoinmunes como anemias
hemolíticas o púrpuras trombocitopénicas. En las anemias hemolíticas autoinmunes la esplenectomía es una

34
alternativa terapeútica cuando la destrucción masiva de hematíes sensibilizados no puede ser controlada por
otros medios terapéuticos.
Historia Clínica. Paciente mujer de 25 años con anemia hemolítica autoinmune en tratamiento con prednisona.
Su hematocrito se ha mantenido persistentemente bajo a pesar de recibir tratamiento con dosis elevadas de
prednisona por lo que ha tenido que indicársele transfusiones sanguíneas. En su frotis de sangre periférica se
observa numerosos esquistocitos (glóbulos rojos fragmentados) y poiquilocitos (glóbulos rojos deformados). Se
le realiza esplenectomía, extirpándose un bazo que pesaba 1000 gramos y tenía una apariencia congestiva,
lográndose controlar la anemia.
Lámina. Se observa una pulpa blanca relativamente disminuida de volumen con respecto a la pulpa roja, con
gran cantidad de hematíes a nivel de los cordones de Billroth y senos medulares. Otro hallazgo importante es la
presencia de normoblastos en los sinusoides esplénicos, lo que refleja la ocurrencia de hematopoyesis
extramedular.

35
PRÁCTICA Nº 5: TRANSTORNOS GENÉTICOS
LÁMINA 21. ESFEROCITOSIS HEREDITARIA
La esferocitosis hereditaria es una forma de anemia hemolítica, genéticamente determinada, ocasionada por
presencia de moléculas anormales de espectrina y anquirina, lo que determina la presencia de defectos en la
membrana de los glóbulos rojos, los que adquieren una forma esférica y pierden su plasticidad. Estos glóbulos
rojos terminan atrapados en el bazo, donde son hemolizados, y tienen una vida media disminuida (menos de 120
días).
Historia Clínica. Paciente varón de 14 años de edad con historia de anemia hemolítica de larga evolución. Ha
recibido múltiples transfusiones sanguíneas. Su madre y uno de sus tres hermanos también presentan la
enfermedad.
Lámina. Extendido de sangre periférica donde se observa que el diámetro de muchos de los glóbulos rojos
observados es menor que el normal y que estos también carecen de la típica palidez central como corresponde a
un disco bicóncavo.
LÁMINA 22. ANEMIA DE CÉLULAS FALCIFORMES
La anemia de células falciformes o anemia drepanocítica es una enfermedad genética autosómica recesiva
resultado de la sustitución de adenina por timina en el gen de la globina beta, ubicada en el cromosoma 11, lo
que conduce a una mutación de ácido glutámico por valina en la posición 6 de la cadena polipeptídica de globina
beta y a la producción de una hemoglobina funcionalmente defectuosa, la hemoglobina S. Debido al cambio de
ese aminoácido, las moléculas de hemoglobina se agregan formando fibras y dándole al hematíe esa forma de
hoz. La transformación del eritrocito se produce cuando no transporta oxígeno, pues con oxihemoglobina, el
glóbulo tiene la forma clásica bicóncava.
La nueva forma provoca dificultad para la circulación de los glóbulos rojos por ello se obstruyen los vasos
sanguíneos y causan síntomas como dolor en las extremidades. Los glóbulos rojos también padecen de una vida
más corta provocando anemia por no ser reemplazados a tiempo. Los glóbulos rojos falciformes no pueden pasar
Lámina 21. Esferocitosis hereditaria
Lámina 22. Anemia de células falciformes
Lámina 23. Hemocromatosis
Lámina 24. Epidermolisis ampollar
Lámina 25. Enfermedad de Gaucher

36
a través de los capilares y las vénulas. Se asocian unos con otros, quedan enganchados debido a su forma
curvada y causan obstrucciones en los vasos. Debido también a sus extremos puntiagudos, pueden llegar a
desgarrar las paredes de los vasos.
La anemia de células falciformes es una enfermedad autosómica recesiva. Los estudios muestran que en las
zonas donde el paludismo por plasmodium falciparum era un problema, especialmente en el Africa, los
individuos que heredaban un solo alelo de la hemoglobina S (y que por tanto eran portadores del rasgo de la
célula falciforme) tenían una ventaja para sobrevivir; a diferencia de los individuos con genes de hemoglobina
normales. A medida que las poblaciones iban migrando de un lugar a otro, la anemia de células falciformes se
extendió a otras zonas del Mediterráneo y de allí al Oriente Medio, y finalmente al hemisferio occidental.
Historia Clínica. Paciente varón de 25 años a quien periódicamente se le realiza transfusiones sanguíneas por
presentar anemia hemolítica.
Lámina. Extendido de lámina periférica de sangre en la que se observa numerosos glóbulos rojos de formas
alargadas, algunos con la característica forma de una hoz.
LÁMINA 23: HEMOCROMATOSIS
Los términos siderosis y hemosiderosis se refieren a la presencia de depósitos de hierro en los tejidos. Las causas
de sobrecarga de hierro pueden ser muchas: hemocromatosis genética, siderosis secundaria a anemia crónica,
sobrecarga neonatal de hierro, transfusión sanguínea, hemólisis, falla renal crónica, porfiria cutánea tarda, entre
otras. Es preferible no utilizar los términos hemosiderosis y hemocromatosis como sinónimos, si no reservar el
término hemocromatosis para referirse a sobrecarga genética o hereditaria de hierro. La hemocromatosis
hereditaria es un transtorno homocigótico recesivo y más del 90% de casos están asociados a mutación en el gen
HFE localizado en el brazo corto del cromosoma 6. Esta mutación produce una disregulación en la homeostasis
de los enterocitos que tiene como consecuencia una absorción incrementada de hierro produciéndose depósitos
de pigmento hemático en hígado, páncreas, miocardio, hipófisis, suprarrenales, tiroides, articulaciones y piel.
Además de los acúmulos de pigmento los pacientes con hemocromatosis desarrollan con frecuencia cirrosis
hepática, fibrosis pancreática, diabetes mellitus, pigmentación cutánea, y tienen una incidencia incrementada de
carcinoma hepatocelular.
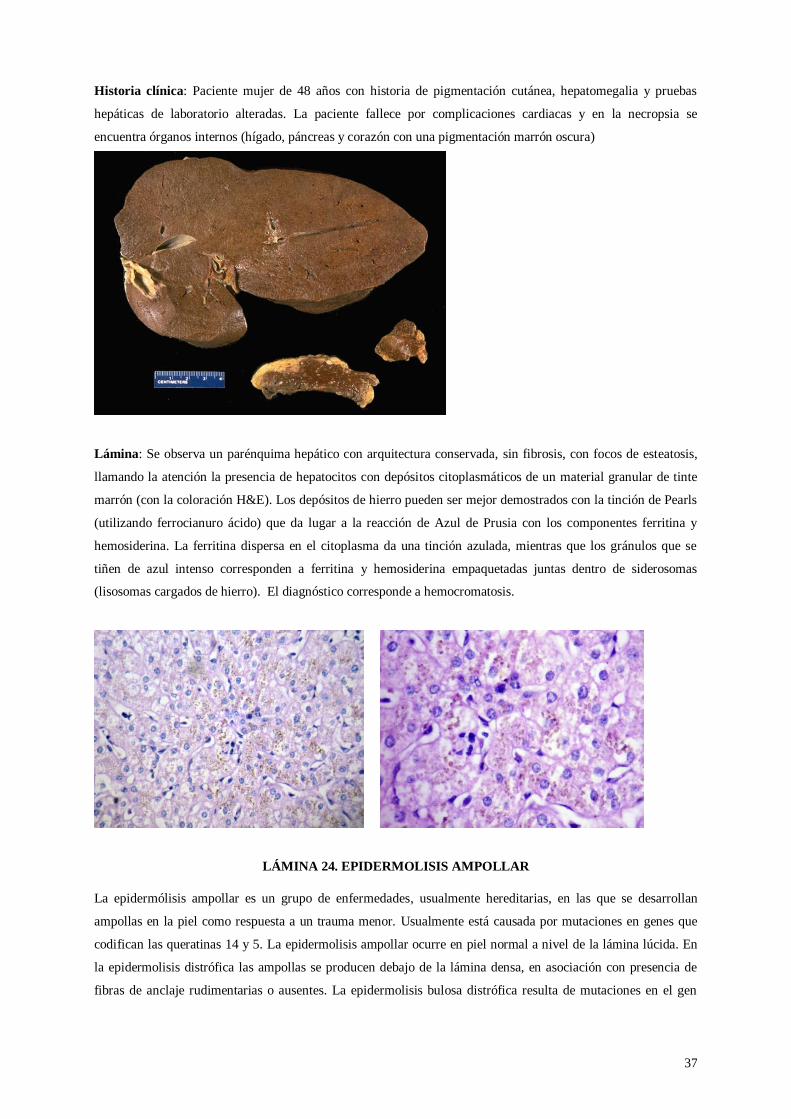
37
Historia clínica: Paciente mujer de 48 años con historia de pigmentación cutánea, hepatomegalia y pruebas
hepáticas de laboratorio alteradas. La paciente fallece por complicaciones cardiacas y en la necropsia se
encuentra órganos internos (hígado, páncreas y corazón con una pigmentación marrón oscura)
Lámina: Se observa un parénquima hepático con arquitectura conservada, sin fibrosis, con focos de esteatosis,
llamando la atención la presencia de hepatocitos con depósitos citoplasmáticos de un material granular de tinte
marrón (con la coloración H&E). Los depósitos de hierro pueden ser mejor demostrados con la tinción de Pearls
(utilizando ferrocianuro ácido) que da lugar a la reacción de Azul de Prusia con los componentes ferritina y
hemosiderina. La ferritina dispersa en el citoplasma da una tinción azulada, mientras que los gránulos que se
tiñen de azul intenso corresponden a ferritina y hemosiderina empaquetadas juntas dentro de siderosomas
(lisosomas cargados de hierro). El diagnóstico corresponde a hemocromatosis.
LÁMINA 24. EPIDERMOLISIS AMPOLLAR
La epidermólisis ampollar es un grupo de enfermedades, usualmente hereditarias, en las que se desarrollan
ampollas en la piel como respuesta a un trauma menor. Usualmente está causada por mutaciones en genes que
codifican las queratinas 14 y 5. La epidermolisis ampollar ocurre en piel normal a nivel de la lámina lúcida. En
la epidermolisis distrófica las ampollas se producen debajo de la lámina densa, en asociación con presencia de
fibras de anclaje rudimentarias o ausentes. La epidermolisis bulosa distrófica resulta de mutaciones en el gen
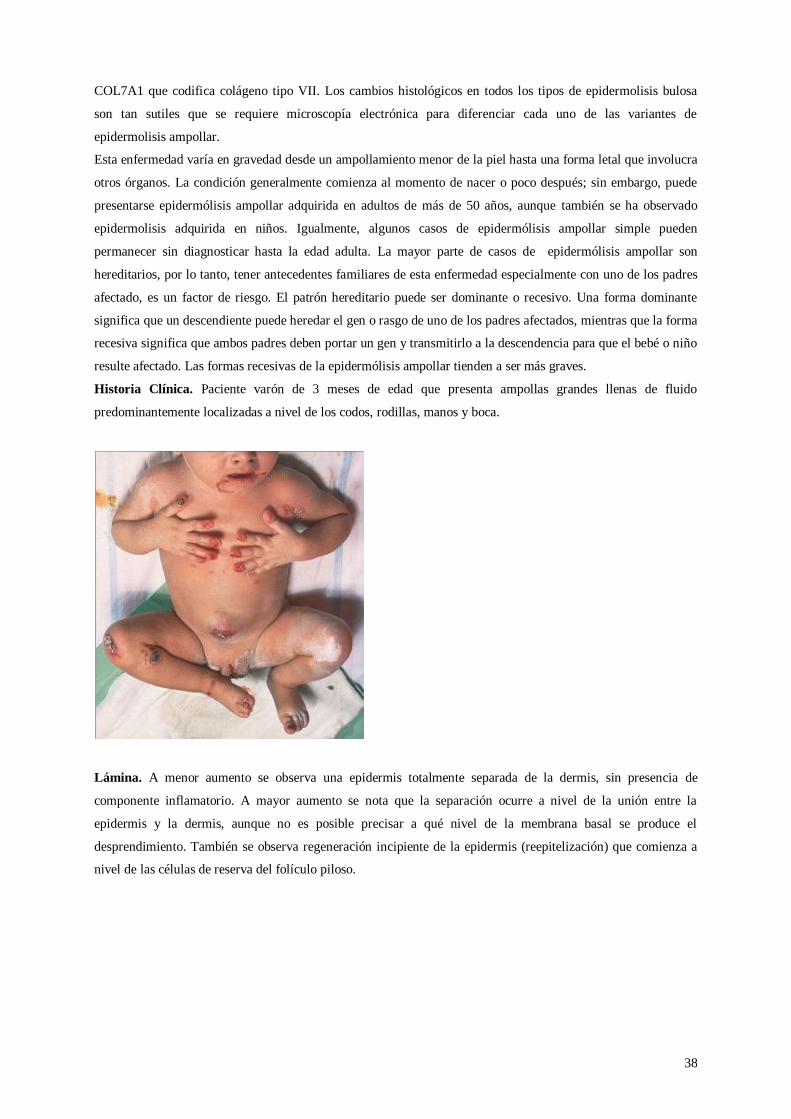
38
COL7A1 que codifica colágeno tipo VII. Los cambios histológicos en todos los tipos de epidermolisis bulosa
son tan sutiles que se requiere microscopía electrónica para diferenciar cada uno de las variantes de
epidermolisis ampollar.
Esta enfermedad varía en gravedad desde un ampollamiento menor de la piel hasta una forma letal que involucra
otros órganos. La condición generalmente comienza al momento de nacer o poco después; sin embargo, puede
presentarse epidermólisis ampollar adquirida en adultos de más de 50 años, aunque también se ha observado
epidermolisis adquirida en niños. Igualmente, algunos casos de epidermólisis ampollar simple pueden
permanecer sin diagnosticar hasta la edad adulta. La mayor parte de casos de epidermólisis ampollar son
hereditarios, por lo tanto, tener antecedentes familiares de esta enfermedad especialmente con uno de los padres
afectado, es un factor de riesgo. El patrón hereditario puede ser dominante o recesivo. Una forma dominante
significa que un descendiente puede heredar el gen o rasgo de uno de los padres afectados, mientras que la forma
recesiva significa que ambos padres deben portar un gen y transmitirlo a la descendencia para que el bebé o niño
resulte afectado. Las formas recesivas de la epidermólisis ampollar tienden a ser más graves.
Historia Clínica. Paciente varón de 3 meses de edad que presenta ampollas grandes llenas de fluido
predominantemente localizadas a nivel de los codos, rodillas, manos y boca.
Lámina. A menor aumento se observa una epidermis totalmente separada de la dermis, sin presencia de
componente inflamatorio. A mayor aumento se nota que la separación ocurre a nivel de la unión entre la
epidermis y la dermis, aunque no es posible precisar a qué nivel de la membrana basal se produce el
desprendimiento. También se observa regeneración incipiente de la epidermis (reepitelización) que comienza a
nivel de las células de reserva del folículo piloso.
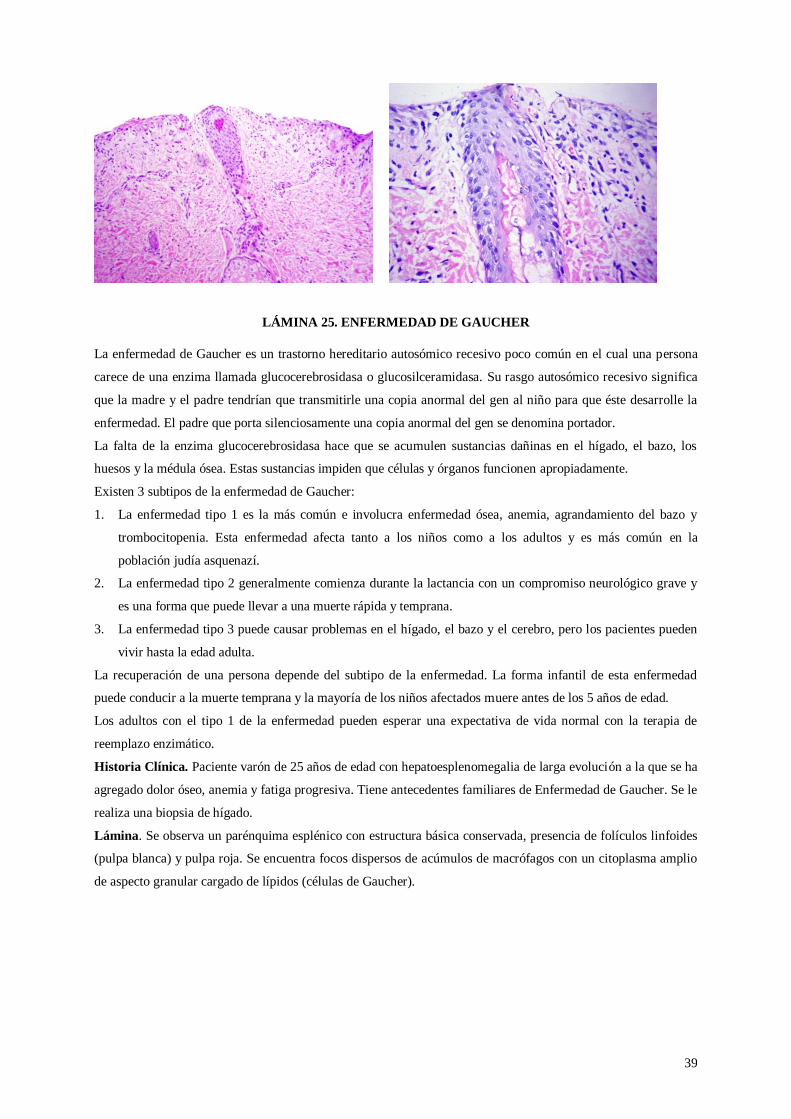
39
LÁMINA 25. ENFERMEDAD DE GAUCHER
La enfermedad de Gaucher es un trastorno hereditario autosómico recesivo poco común en el cual una persona
carece de una enzima llamada glucocerebrosidasa o glucosilceramidasa. Su rasgo autosómico recesivo significa
que la madre y el padre tendrían que transmitirle una copia anormal del gen al niño para que éste desarrolle la
enfermedad. El padre que porta silenciosamente una copia anormal del gen se denomina portador.
La falta de la enzima glucocerebrosidasa hace que se acumulen sustancias dañinas en el hígado, el bazo, los
huesos y la médula ósea. Estas sustancias impiden que células y órganos funcionen apropiadamente.
Existen 3 subtipos de la enfermedad de Gaucher:
1. La enfermedad tipo 1 es la más común e involucra enfermedad ósea, anemia, agrandamiento del bazo y
trombocitopenia. Esta enfermedad afecta tanto a los niños como a los adultos y es más común en la
población judía asquenazí.
2. La enfermedad tipo 2 generalmente comienza durante la lactancia con un compromiso neurológico grave y
es una forma que puede llevar a una muerte rápida y temprana.
3. La enfermedad tipo 3 puede causar problemas en el hígado, el bazo y el cerebro, pero los pacientes pueden
vivir hasta la edad adulta.
La recuperación de una persona depende del subtipo de la enfermedad. La forma infantil de esta enfermedad
puede conducir a la muerte temprana y la mayoría de los niños afectados muere antes de los 5 años de edad.
Los adultos con el tipo 1 de la enfermedad pueden esperar una expectativa de vida normal con la terapia de
reemplazo enzimático.
Historia Clínica. Paciente varón de 25 años de edad con hepatoesplenomegalia de larga evolución a la que se ha
agregado dolor óseo, anemia y fatiga progresiva. Tiene antecedentes familiares de Enfermedad de Gaucher. Se le
realiza una biopsia de hígado.
Lámina. Se observa un parénquima esplénico con estructura básica conservada, presencia de folículos linfoides
(pulpa blanca) y pulpa roja. Se encuentra focos dispersos de acúmulos de macrófagos con un citoplasma amplio
de aspecto granular cargado de lípidos (células de Gaucher).

40

41
PRÁCTICA N° 6: NEOPLASIAS BENIGNAS
En términos generales, neoplasia es un nuevo crecimiento de tejido, de naturaleza autónoma, que sigue sus
propias leyes de crecimiento, sin tener en cuenta el beneficio del organismo en su conjunto y que crece a
expensas del cuerpo y no para su beneficio. Con algunas excepciones las neoplasias pueden clasificarse como
benignas o malignas. Algunas veces no es posible, en base a criterios morfológicos ni con ayuda de técnicas
especiales, definir el potencial maligno de una neoplasia. Estas neoplasias son llamadas de tipo borderline o en el
límite de la malignidad.
Las neoplasias benignas suelen tener un crecimiento expansivo local, y la histología se asemeja a la célula
original, con pocas mitosis, índice núcleo/citoplasma normal o ligeramente aumentado, y las células son
uniformes en todo el tumor.
Las neoplasias según su evolución se clasifican en benignas y malignas.
Las neoplasias benignas producen sólo alteraciones locales, generalmente de orden mecánico como en el
leiomioma uterino. Estas rara vez producen la muerte, aunque dependiendo de factores topográficos o
funcionales la neoplasia misma puede ser letal. Ejemplos: meningioma que causa compresión del cerebro,
adenoma paratiroideo con hipercalcemia.
Caracteres generales de las neoplasias benignas
Crecimiento lento (meses o años)
Crecimiento expansivo
Tumores redondeados, a veces encapsulados, bien delimitados. Pueden ser extirpados quirúrgicamente por
completo, sin que vuelvan a aparecer.
Células típicas del tejido en que se originan, es decir, células muy bien diferenciadas
Mitosis escasas o ausentes
Lámina 26. MÚSCULO LISO: LEIOMIOMA
En términos generales la distribución de los tumores benignos de músculo liso es paralela a la distribución del
músculo liso en los tejidos corporales. En un estudio de 7748 leiomiomas (Farman, S Afr Med J 1974; 48: 1214)
reportó que el 95% ocurrieron en el tracto genital femenino, siendo los demás localizados, en orden de
frecuencia, en la piel, tracto gastrointestinal y vejiga. Se observa leiomiomas en aproximadamente el 77% de
especímenes de histerectomía independientemente de la indicación quirúrgica, por tal motivo se estima que estos
son los tumores humanos más frecuentes. Afortunadamente, sólo alrededor del 25% de mujeres en edad
reproductiva con leiomiomas son sintomáticas. La mayor parte de los leiomiomas se localizan en el útero y los
síntomas que producen son atribuibles a su localización dentro de la pared uterina. Los leiomiomas submucosos
suelen manifestarse con sangrado uterino anormal o pueden prolapsar a través de la cavidad uterina. Los
leiomiomas intramurales o subserosos suelen dar síntomas de dolor o presión pélvica. A veces los leiomiomas
Lámina 26. Leiomioma
Lámina 27. Neurofibroma
Lámina 28. Hemangioma capilar
Lámina 29. Adenoma velloso
Lámina 30. Meningioma
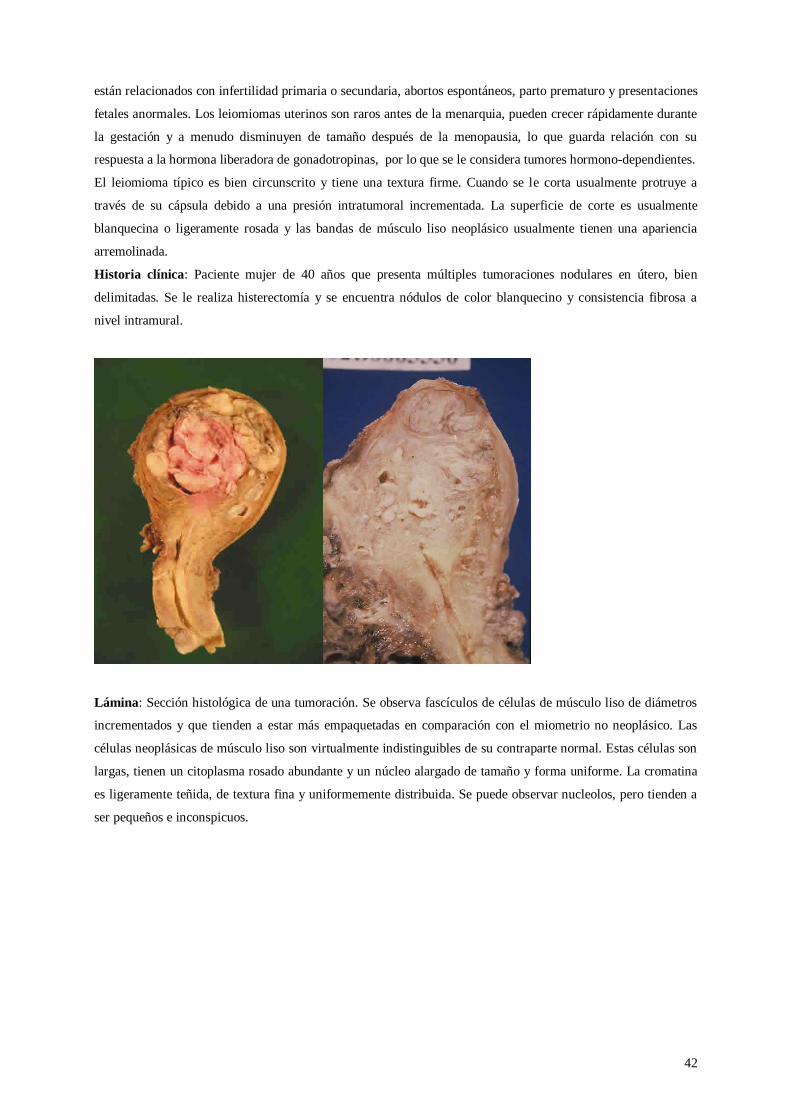
42
están relacionados con infertilidad primaria o secundaria, abortos espontáneos, parto prematuro y presentaciones
fetales anormales. Los leiomiomas uterinos son raros antes de la menarquia, pueden crecer rápidamente durante
la gestación y a menudo disminuyen de tamaño después de la menopausia, lo que guarda relación con su
respuesta a la hormona liberadora de gonadotropinas, por lo que se le considera tumores hormono-dependientes.
El leiomioma típico es bien circunscrito y tiene una textura firme. Cuando se le corta usualmente protruye a
través de su cápsula debido a una presión intratumoral incrementada. La superficie de corte es usualmente
blanquecina o ligeramente rosada y las bandas de músculo liso neoplásico usualmente tienen una apariencia
arremolinada.
Historia clínica: Paciente mujer de 40 años que presenta múltiples tumoraciones nodulares en útero, bien
delimitadas. Se le realiza histerectomía y se encuentra nódulos de color blanquecino y consistencia fibrosa a
nivel intramural.
Lámina: Sección histológica de una tumoración. Se observa fascículos de células de músculo liso de diámetros
incrementados y que tienden a estar más empaquetadas en comparación con el miometrio no neoplásico. Las
células neoplásicas de músculo liso son virtualmente indistinguibles de su contraparte normal. Estas células son
largas, tienen un citoplasma rosado abundante y un núcleo alargado de tamaño y forma uniforme. La cromatina
es ligeramente teñida, de textura fina y uniformemente distribuida. Se puede observar nucleolos, pero tienden a
ser pequeños e inconspicuos.
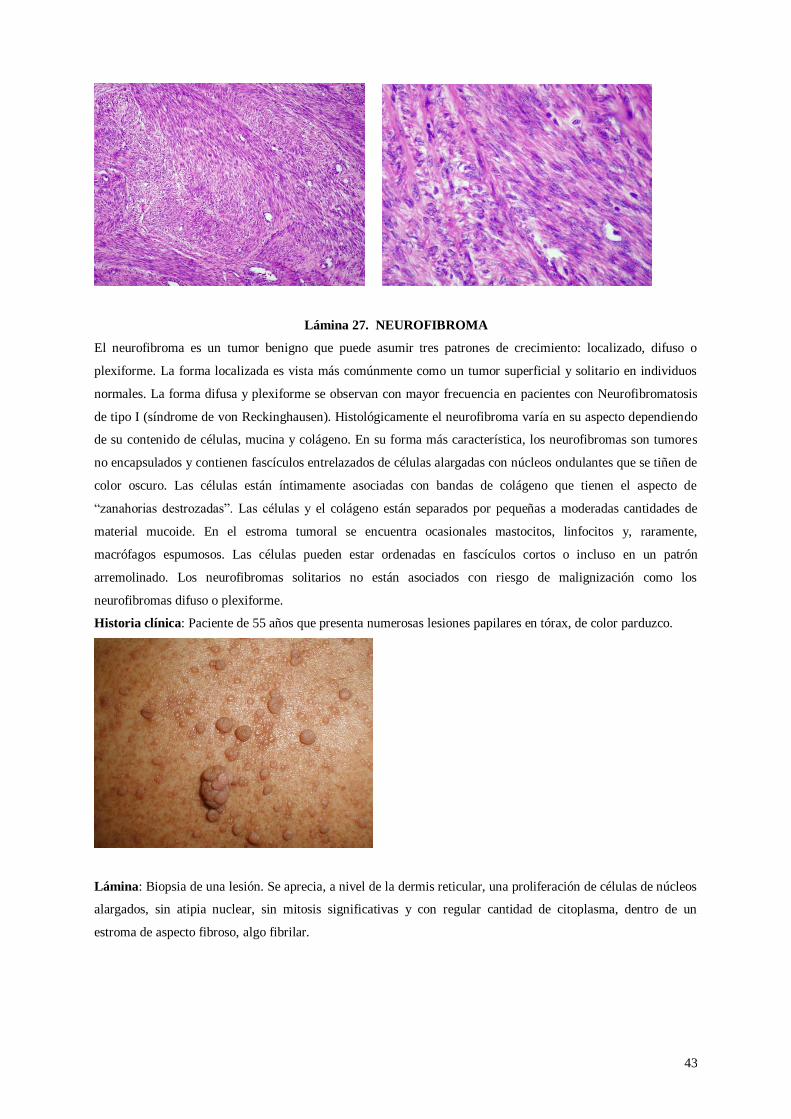
43
Lámina 27. NEUROFIBROMA
El neurofibroma es un tumor benigno que puede asumir tres patrones de crecimiento: localizado, difuso o
plexiforme. La forma localizada es vista más comúnmente como un tumor superficial y solitario en individuos
normales. La forma difusa y plexiforme se observan con mayor frecuencia en pacientes con Neurofibromatosis
de tipo I (síndrome de von Reckinghausen). Histológicamente el neurofibroma varía en su aspecto dependiendo
de su contenido de células, mucina y colágeno. En su forma más característica, los neurofibromas son tumores
no encapsulados y contienen fascículos entrelazados de células alargadas con núcleos ondulantes que se tiñen de
color oscuro. Las células están íntimamente asociadas con bandas de colágeno que tienen el aspecto de
“zanahorias destrozadas”. Las células y el colágeno están separados por pequeñas a moderadas cantidades de
material mucoide. En el estroma tumoral se encuentra ocasionales mastocitos, linfocitos y, raramente,
macrófagos espumosos. Las células pueden estar ordenadas en fascículos cortos o incluso en un patrón
arremolinado. Los neurofibromas solitarios no están asociados con riesgo de malignización como los
neurofibromas difuso o plexiforme.
Historia clínica: Paciente de 55 años que presenta numerosas lesiones papilares en tórax, de color parduzco.
Lámina: Biopsia de una lesión. Se aprecia, a nivel de la dermis reticular, una proliferación de células de núcleos
alargados, sin atipia nuclear, sin mitosis significativas y con regular cantidad de citoplasma, dentro de un
estroma de aspecto fibroso, algo fibrilar.
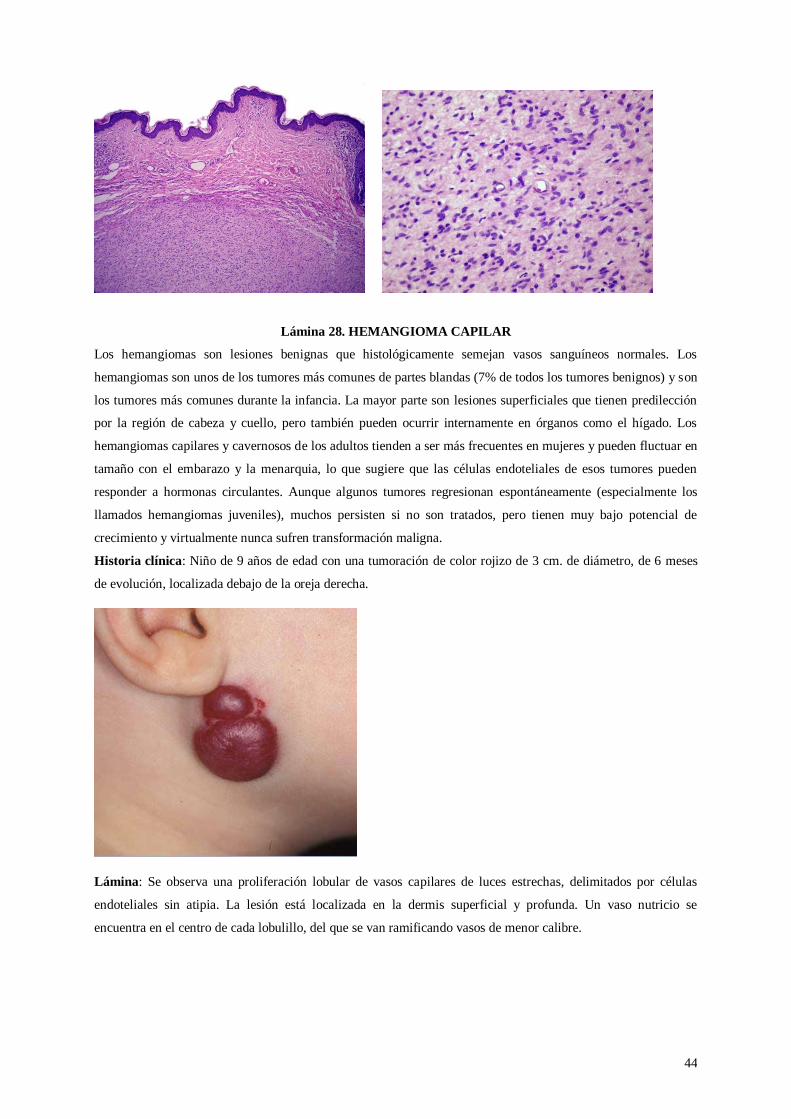
44
Lámina 28. HEMANGIOMA CAPILAR
Los hemangiomas son lesiones benignas que histológicamente semejan vasos sanguíneos normales. Los
hemangiomas son unos de los tumores más comunes de partes blandas (7% de todos los tumores benignos) y son
los tumores más comunes durante la infancia. La mayor parte son lesiones superficiales que tienen predilección
por la región de cabeza y cuello, pero también pueden ocurrir internamente en órganos como el hígado. Los
hemangiomas capilares y cavernosos de los adultos tienden a ser más frecuentes en mujeres y pueden fluctuar en
tamaño con el embarazo y la menarquia, lo que sugiere que las células endoteliales de esos tumores pueden
responder a hormonas circulantes. Aunque algunos tumores regresionan espontáneamente (especialmente los
llamados hemangiomas juveniles), muchos persisten si no son tratados, pero tienen muy bajo potencial de
crecimiento y virtualmente nunca sufren transformación maligna.
Historia clínica: Niño de 9 años de edad con una tumoración de color rojizo de 3 cm. de diámetro, de 6 meses
de evolución, localizada debajo de la oreja derecha.
Lámina: Se observa una proliferación lobular de vasos capilares de luces estrechas, delimitados por células
endoteliales sin atipia. La lesión está localizada en la dermis superficial y profunda. Un vaso nutricio se
encuentra en el centro de cada lobulillo, del que se van ramificando vasos de menor calibre.

45
Lámina 29. ADENOMA VELLOSO
Los adenomas del intestino grueso y recto son lesiones comunes que casi siempre son asintomáticas. La mayor
parte de pacientes detectados se presentan con sangrado en heces evidente u oculto. La importancia clínica de los
adenomas está casi enteramente relacionada con su bien establecida naturaleza premaligna. En general, la
prevalencia de adenomas se incrementa dramáticamente con la edad. Alrededor de la quinta década de vida
aproximadamente el 12% de individuos tiene adenomas, de los cuales el 25% son considerados lesiones de alto
riesgo. La probabilidad de desarrollar adenomas está altamente relacionada por su historia familiar y por una
variedad de factores nutricionales. Los pólipos adenomatosos del colon pueden ser clasificados como
convencionales (tubulares, tubulovellosos o vellosos), aserrados o planos. Un adenoma se define como una
lesión displásica y clonal de epitelio que puede ser sésil o pediculada y que puede tener ulceración superficial. La
displasia de bajo grado en un adenoma se define por la presencia de criptas arquitecturalmente no complejas que
contienen núcleos pseudoestratificados o parcialmente estratificados al punto que el núcleo celular sólo alcanza
la mitad inferior del citoplasma celular. Puede encontrarse actividad mitótica, pero mitosis atípicas, pérdida
significativa de polaridad o pleomorfismo son mínimos. Las criptas están arregladas en una configuración
paralela, sin estar tan pegadas unas con otras, sin patrón cribiforme ni gemaciones complejas. La displasia de
alto grado se define como una marcada pseudoestratificación o estratificación de células neoplásicas cuyos
núcleos se extienden por encima de la mitad luminal de las células. Usualmente presentan marcado
pleomorfismo, actividad mitótica incrementada, mitosis atípicas y pérdida de polaridad nuclear. Con la
progresión de la displasia las glándulas pierden su configuración ordenada y se tornan más irregulares y
complejas. Adicionalmente, los núcleos aumentan de tamaño y pueden contener nucleolos prominentes, la
relación núcleo/citoplasma de la célula se incrementa y la pérdida de polaridad se vuelve marcada. En este punto
la denominación de carcinoma in-situ reemplaza a la de displasia de alto grado.
Historia clínica: Paciente de 67 años con tumoración rectal exofítica que mide 4 x 3 cm.

46
Lámina: Biopsia de la tumoración. Se observa una lesión arborescente, en forma de pólipo pediculado, que
ocupa una amplia zona de la mucosa. La lesión está formada por proyecciones epiteliales papilares revestidas por
células epiteliales con cambios displásicos (núcleos alargados, hipercromáticos y superpuestos) y presencia de
mitosis frecuentes. Existe tejido conectivo entre el revestimiento epitelial. A nivel del pedículo se observa la
transición entre el epitelio displásico y la mucosa rectal normal.
Lámina 30. MENINGIOMA
Los meningiomas son generalmente tumores benignos de crecimiento lento que suelen estar unidos a la
duramadre y están compuestos por células meningoteliales (aracnoideas) neoplásicas. Los meningiomas fueron
los primeros tumores sólidos en que se reconoció la presencia de alteraciones citogenéticas. La alteración
genética más frecuente en estos tumores es la pérdida del cromosoma 22. Típicamente se presentan en adultos y
son más frecuentes en mujeres. Se estima que los meningiomas constituyen entre el 13 a 26% de los tumores
intracraneales primarios en los Estados Unidos. Muchos meningiomas pequeños no son detectados en vida. Tales
meningiomas incidentales son ahora detectados con mayor frecuencia debido al uso de tomografía
computarizada y resonancia magnética. Los meningiomas son a menudo múltiples en pacientes con
neurofibromatosis tipo 2 (NF2) y familias sin mutaciones en NF2 pero con predisposición hereditaria a
meningiomas. A pesar que los meningiomas son generalmente benignos, un 5% a 7% son considerados atípicos
(localmente infiltrativos y recidivantes) y un 1% a 3% son anaplásicos o malignos (potencialmente
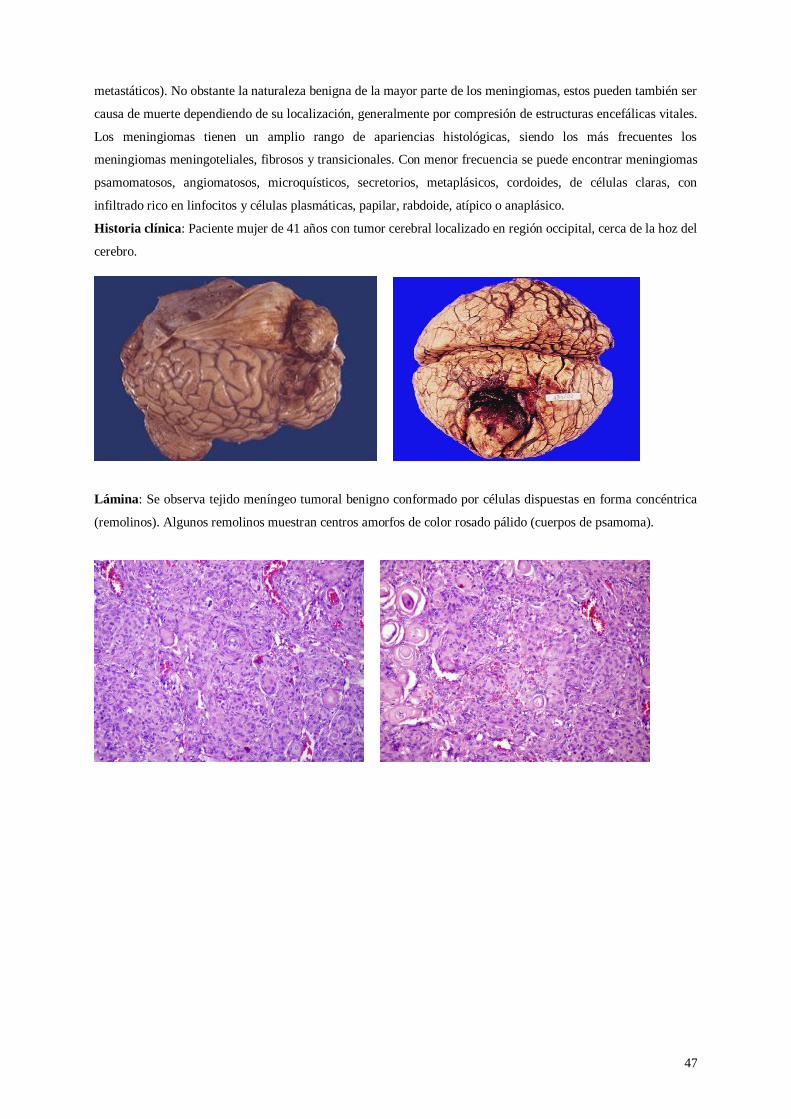
47
metastáticos). No obstante la naturaleza benigna de la mayor parte de los meningiomas, estos pueden también ser
causa de muerte dependiendo de su localización, generalmente por compresión de estructuras encefálicas vitales.
Los meningiomas tienen un amplio rango de apariencias histológicas, siendo los más frecuentes los
meningiomas meningoteliales, fibrosos y transicionales. Con menor frecuencia se puede encontrar meningiomas
psamomatosos, angiomatosos, microquísticos, secretorios, metaplásicos, cordoides, de células claras, con
infiltrado rico en linfocitos y células plasmáticas, papilar, rabdoide, atípico o anaplásico.
Historia clínica: Paciente mujer de 41 años con tumor cerebral localizado en región occipital, cerca de la hoz del
cerebro.
Lámina: Se observa tejido meníngeo tumoral benigno conformado por células dispuestas en forma concéntrica
(remolinos). Algunos remolinos muestran centros amorfos de color rosado pálido (cuerpos de psamoma).

48
PRÁCTICA N° 7: NEOPLASIAS MALIGNAS
El término neoplasia maligna ha sido considerado durante mucho tiempo sinónimo de cáncer. En condiciones
normales, la proliferación celular es un proceso cuidadosamente regulado desde el momento en que el huevo se
fecunda hasta el final de la vida; sin embargo, a veces esta capacidad reguladora se pierde y sobreviene el cáncer.
Desde este punto de vista, el cáncer puede considerarse como un estado de desequilibrio en el cual la velocidad
de crecimiento de un tejido excede notablemente sus necesidades de sostén. Por consiguiente, un cáncer deriva
de las propias células del organismo, y han adquirido funciones nocivas al extremo de que el crecimiento
progresivo e incesante domina su actividad. A medida que estas células crecen, invaden y destruyen el tejido
normal, se diseminan a distancia y acaban con la integridad del organismo.
La palabra cáncer deriva del latín y significa “cangrejo”. Se aplicó por primera vez al cáncer de mama por su
semejanza en su forma de infiltración con la forma del cangrejo, cuyas tenazas y patas se proyectaban como
rayos a partir de un cuerpo central y representaban el crecimiento invasor hacia los tejidos circundantes. Los
documentos médicos más antiguos describiendo esta enfermedad datan de 1200 años antes de Cristo y fueron
encontrados en Egipto. Estudiando las causas externas del cáncer, Sir Percival Pott observó en 1775 una elevada
frecuencia de carcinoma de escroto entre limpiadores de chimeneas y formuló las relaciones causales entre
profesión y cáncer. Los conceptos modernos de esta enfermedad tienen su origen en los trabajos de Johannes
Müller, en 1838, quien observó que las neoplasias estaban compuestas por tejidos celulares. Veinte años
después, Virchow dio un nuevo impulso a esta idea y describió las alteraciones celulares características del
cáncer. En 1889 Hanau transplantó un cáncer de una rata a otra. La importancia de los factores genéticos
comenzó a advertirse cuando Tyzzer produjo en 1907 varias cepas de ratas de gran pureza y observó notables
diferencias en la frecuencia de cáncer mamario entre ellas. En 1911 Peyton Rous describió un virus transmisible
que producía cáncer en pollos. En 1916 se descubrió el primer agente carcinogénico cuando Yamagawa e
Ichikawa hicieron la observación de que cuando las orejas de los conejos se untaban con alquitrán de hulla
durante mucho tiempo se producían tumores, algunos de los cuales terminaban siendo cancerosos.
La evolución del conocimiento del cáncer es notable hasta nuestros días. Quizás el avance de mayor
trascendencia es el hecho de reconocer que el cáncer es una enfermedad de naturaleza genética caracterizada por
una acumulación progresiva de mutaciones, muchas de las cuales se producen aún antes de demostrarse una
alteración observable microscópicamente. (Siendo algunas mutaciones características de la iniciación y/o
progresión de algunos tipos de cánceres).
Con excepciones, las características generales de las neoplasias malignas son las siguientes:
Anaplasia
Pleomorfismo
Hipercromasia
Relación núcleo/citoplasma aumentada
Lámina 31. Carcinoma epidermoide
Lámina 32. Adenocarcinoma de ovario
Lámina 33. Linfoma no Hodgkin
Lámina 34. Linfoma de Hodgkin
Lámina 35. Seminoma

49
Relación nucleolo/núcleo aumentada
Mitosis anormales (cualitativamente)
Mitosis anormales (cuantitativamente)
Función anormal
Organización alterada
o Pérdida de la estratificación normal
o Pérdida de polaridad
o Pobre delimitación
o Acumulación de productos de secreción
Invasión destructiva del tejido circundante
Metástasis a distancia
Tendencia a sufrir necrosis
Provoca reacción del tejido circundante (inflamación, fibrosis)
Lámina 31. CARCINOMA EPIDERMOIDE
El carcinoma epidermoide, también llamado carcinoma escamoso, es una neoplasia maligna que se origina en el
epitelio poliestratificado plano. Este epitelio recubre normalmente la piel, donde es queratinizado, y superficies
mucosas (boca, parte de la faringe, esófago, ano, vagina, exocérvix uterino y pene), por lo que se deduce que los
carcinomas epidermoides tendrán similares localizaciones. Una excepción a esta regla es la presencia de
carcinoma epidermoide de pulmón, el que se asume que se origina de una metaplasia escamosa previa del
epitelio respiratorio bronquial, la que posteriormente sufre la transformación maligna. En la piel y mucosas los
carcinomas epidermoides tienen diferentes formas clínicas: Enfermedad de Bowen (carcinoma epidermoide in
situ que se presenta en forma de una placa), eritroplasia de Queyrat (carcinoma epidermoide in situ del pene y la
vulva), carcinoma verrucoso (un tipo de carcinoma epidermoide bien diferenciado generalmente asociado a
infección por papilomavirus humano). La relación entre infección por papilomavirus humano y los carcinomas
epidermoides en regiones genitales es muy conocida y su investigación extensa ha conducido a la producción de
la vacuna contra el papilomavirus humano, cuyo uso masivo conduciría a reducir dramáticamente la incidencia
de cáncer de cuello uterino.
Historia clínica: Paciente varón de 56 años de edad, fumador crónico, que presenta tos, dificultad respiratoria
progresiva y una masa perihiliar en la tomografía de tórax. Se realiza una biopsia a cielo abierto.

50
Lámina: Sección histológica de la lesión donde se aprecia una proliferación epitelial escamosa con desorden en
el patrón arquitectural, presencia de mitosis atípicas e invasión del tejido adyacente. Se observan perlas córneas
(nidos de queratina).
Lámina 32. ADENOCARCINOMA DE OVARIO
En términos generales, las neoplasias (benignas y malignas) de ovario se pueden clasificar en tres grandes
grupos: los tumores epiteliales (cistoadenomas y cistoadenocarcinomas), que pueden ser serosos o mucinosos;
los tumores germinativos (teratoma, carcinoma embrionario, coriocarcinoma, tumor del seno endodermal,
disgerminoma); y los tumores de los cordones sexuales (tecoma, fibrotecoma, tumor de células de la granulosa).
Los tumores más frecuentes en mujeres menores de 20 años son los teratomas, que son tumores germinativos,
generalmente benignos, que demuestran diferenciación de tipo mesodérmico y endodérmico, principalmente, y
que suelen contener diferentes elementos como pelos, dientes, queratina, tejido adiposo, etc. En mujeres de
mayor edad son más frecuentemente detectados tumores epiteliales, generalmente quísticos y usualmente
benignos, que representan una proliferación anormal de epitelio de tipo seroso o mucinoso. En estos tumores,
desde el punto de vista macroscópico, los mejores indicadores de malignidad son la complejidad del tumor, el
predominio de áreas sólidas sobre áreas quísticas y la evidencia de infiltración de la pared del tumor. Desde el
punto de vista microscópico, la determinación de la malignidad de un tumor epitelial de ovario se basa en la
presencia de estratificación de las células neoplásica, el grado de atipia celular y la invasión del estroma ovárico;
aunque a veces algunos de estos tumores no pueden ser definitivamente catalogados como benignos o malignos,
habiéndose acuñado la denominación de tumor de malignidad incierta, tumor limítrofe o “borderline” para
clasificarlos adecuadamente.
Historia clínica: Paciente mujer de 45 años de edad que presenta tumoración quística de ovario derecho que
mide 15 x 15 cm. Tiene niveles elevados de antígeno carcinoembrionario en suero (CEA) y en su ecografía se
observó un quiste de estructura interna compleja, con áreas sólidas. Al examen macroscópico se encuentra una
tumoración quística conteniendo líquido seroso, con numerosas trabeculaciones en su interior y áreas sólidas que
constituyen el 20% de todo su volumen.

51
Lámina: A menor aumento se observa una proliferación de glándulas serosas en un patrón desordenado
invadiendo el estroma ovárico. A mayor aumento se observa que las células que tapizan las glándulas tienen
núcleos hipercromáticos, con superposición y estratificación de nuclear.
Lámina 33. LINFOMA NO HODGKIN
Bajo la denominación de linfoma no Hodgkin se agrupa un complejo grupo de neoplasias linfoides que tienen
como denominador común el no ser de tipo Hodgkin (descrito más adelante). Estas neoplasias pueden ser sólidas
o de tipo leucémico; comprometen generalmente ganglios linfáticos, aunque pueden afectar virtualmente
cualquier órgano de la economía, no siendo raro encontrarlos en estómago, piel, intestinos o amígdalas; pueden
tener un curso clínico muy agresivo o evolucionar lentamente a lo largo de muchos años y generalmente están
constituidas por un solo tipo celular formando una masa tumoral, aunque a veces las células malignas se
encuentran dispersas en medio de un infiltrado inflamatorio prominente, siendo difícil detectarlas. La
clasificación general de los linfomas no Hodgkin tiene en cuenta en primer lugar su célula de origen: linfocito T,
B o NK: y el órgano primariamente afectado. A partir de estos dos parámetros se ha diseñado una clasificación
que, teniendo en cuenta aspectos clínicos, la inmunohistoquímica del tumor, la citometría de flujo y la genética,
nos permiten clasificar adecuadamente a la mayoría de estas neoplasias. En los ganglios linfáticos la mayor parte
de linfomas de células B se originan en los centros germinativos, en la zona marginal o en las células del manto,
mientras que los linfomas de células T suelen originarse en la zona paracortical y medular. Aunque en términos
generales se considera que los linfomas de células B tienen mejor pronóstico que los de células T y, a su vez,
estos mejor pronóstico que los de células NK. La entidad clínicopatológica más representativa dentro de los
Linfomas no Hodgkin de ganglios linfáticos es el Linfoma Centrofolicular, que es el Linfoma de células B más
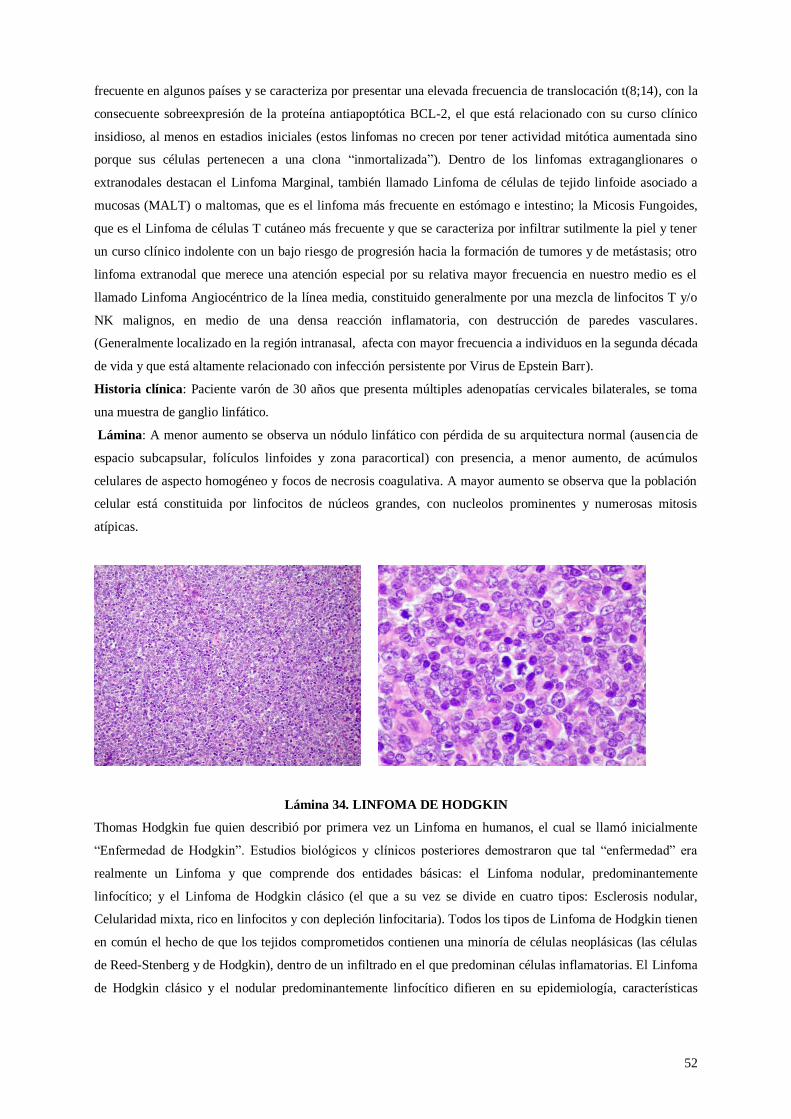
52
frecuente en algunos países y se caracteriza por presentar una elevada frecuencia de translocación t(8;14), con la
consecuente sobreexpresión de la proteína antiapoptótica BCL-2, el que está relacionado con su curso clínico
insidioso, al menos en estadios iniciales (estos linfomas no crecen por tener actividad mitótica aumentada sino
porque sus células pertenecen a una clona “inmortalizada”). Dentro de los linfomas extraganglionares o
extranodales destacan el Linfoma Marginal, también llamado Linfoma de células de tejido linfoide asociado a
mucosas (MALT) o maltomas, que es el linfoma más frecuente en estómago e intestino; la Micosis Fungoides,
que es el Linfoma de células T cutáneo más frecuente y que se caracteriza por infiltrar sutilmente la piel y tener
un curso clínico indolente con un bajo riesgo de progresión hacia la formación de tumores y de metástasis; otro
linfoma extranodal que merece una atención especial por su relativa mayor frecuencia en nuestro medio es el
llamado Linfoma Angiocéntrico de la línea media, constituido generalmente por una mezcla de linfocitos T y/o
NK malignos, en medio de una densa reacción inflamatoria, con destrucción de paredes vasculares.
(Generalmente localizado en la región intranasal, afecta con mayor frecuencia a individuos en la segunda década
de vida y que está altamente relacionado con infección persistente por Virus de Epstein Barr).
Historia clínica: Paciente varón de 30 años que presenta múltiples adenopatías cervicales bilaterales, se toma
una muestra de ganglio linfático.
Lámina: A menor aumento se observa un nódulo linfático con pérdida de su arquitectura normal (ausencia de
espacio subcapsular, folículos linfoides y zona paracortical) con presencia, a menor aumento, de acúmulos
celulares de aspecto homogéneo y focos de necrosis coagulativa. A mayor aumento se observa que la población
celular está constituida por linfocitos de núcleos grandes, con nucleolos prominentes y numerosas mitosis
atípicas.
Lámina 34. LINFOMA DE HODGKIN
Thomas Hodgkin fue quien describió por primera vez un Linfoma en humanos, el cual se llamó inicialmente
“Enfermedad de Hodgkin”. Estudios biológicos y clínicos posteriores demostraron que tal “enfermedad” era
realmente un Linfoma y que comprende dos entidades básicas: el Linfoma nodular, predominantemente
linfocítico; y el Linfoma de Hodgkin clásico (el que a su vez se divide en cuatro tipos: Esclerosis nodular,
Celularidad mixta, rico en linfocitos y con depleción linfocitaria). Todos los tipos de Linfoma de Hodgkin tienen
en común el hecho de que los tejidos comprometidos contienen una minoría de células neoplásicas (las células
de Reed-Stenberg y de Hodgkin), dentro de un infiltrado en el que predominan células inflamatorias. El Linfoma
de Hodgkin clásico y el nodular predominantemente linfocítico difieren en su epidemiología, características

53
clínicas, inmunofenotipo, genética, asociación con infección por virus de Epstein-Barr e historia natural. El
pronóstico en estas neoplasias suele ser bueno, incluso con curación.
Historia clínica: Paciente varón de 50 años con conglomerados ganglionares cervicales de hasta 5 cm. de
diámetro, la radiografía de mediastino muestra masas mediastinales de hasta 10 cm. El paciente ha
experimentado pérdida de peso, prurito y fiebre en los últimos dos meses. Se toma una biopsia del conglomerado
ganglionar cervical.
Lámina: La característica de esta neoplasia es que muestra un infiltrado mixto en el que se encuentran
numerosas células inflamatorias (linfocitos, neutrófilos, eosinófilos y macrófagos), con presencia de cantidades
variables de células neoplásicas binucleadas con nucleolos rojos prominentes (dando la impresión de "ojos de
buho"), llamadas células de Reed-Sternberg.
Lámina 35. SEMINOMA
Seminoma es para el testículo lo que disgerminoma es para el ovario. Estas son neoplasias originadas en el
epitelio germinativo de las gónadas. Por razones desconocidas los seminomas constituyen un 30 a 40% de todos
los tumores testiculares, mientras que los disgerminomas sólo representan el 1% de todos los tumores ováricos.
Existen dos variantes principales de seminomas, el clásico y el espermatocítico. El seminoma clásico comprende
aproximadamente el 93% de todos los seminomas y tiene una apariencia macroscópica característica: es
usualmente de tamaño moderado, sólido, homogéneo, de color amarillo claro y puede contener áreas
circunscritas de necrosis.
Historia Clínica. Paciente varón de 38 años con tumoración en testículo derecho de 3 meses de evolución, que
mide 4 cm. de diámetro.
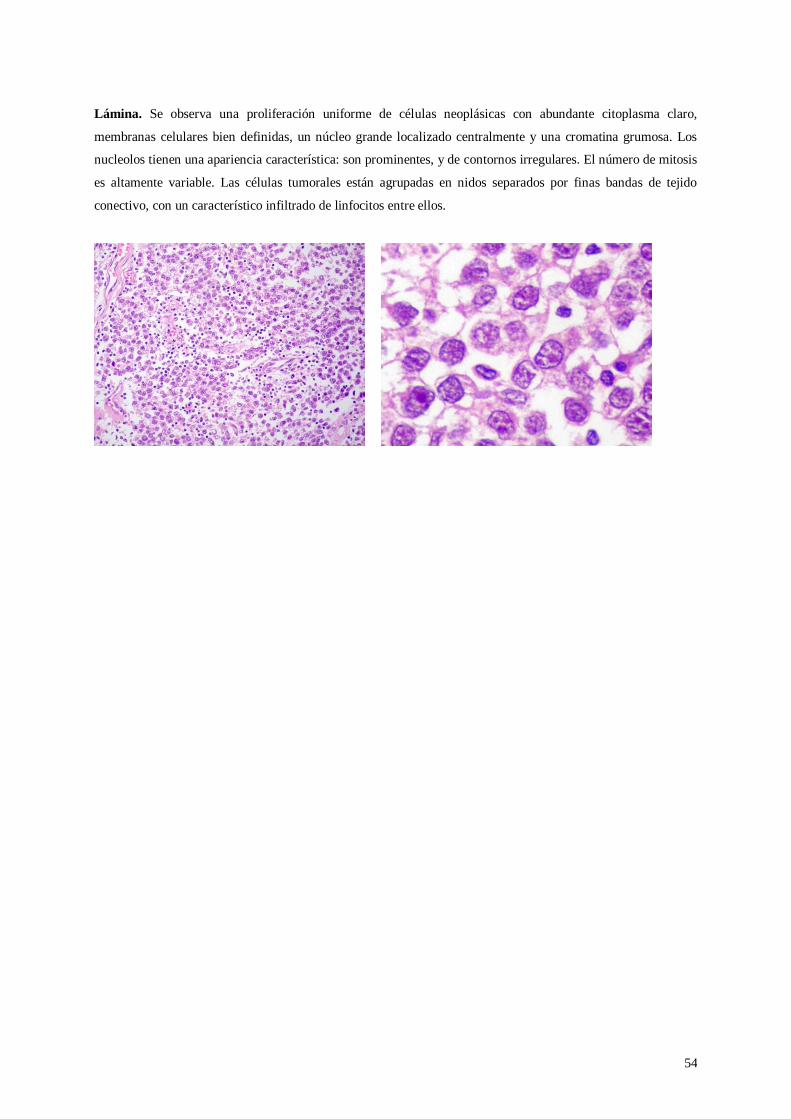
54
Lámina. Se observa una proliferación uniforme de células neoplásicas con abundante citoplasma claro,
membranas celulares bien definidas, un núcleo grande localizado centralmente y una cromatina grumosa. Los
nucleolos tienen una apariencia característica: son prominentes, y de contornos irregulares. El número de mitosis
es altamente variable. Las células tumorales están agrupadas en nidos separados por finas bandas de tejido
conectivo, con un característico infiltrado de linfocitos entre ellos.

55
Práctica Nº 8. ENFERMEDADES INFECCIOSAS
Una de las causas más importantes de inflamación son las infecciones. Las infecciones pueden ser producidas
por bacterias, virus, hongos o parásitos. Los agentes infecciosos no sólo proliferan en el interior del cuerpo,
aumentando sus fuerzas, sino que se adaptan al medio metabólico cambiante proporcionado por el huésped. En
consecuencia, la inflamación que caracteriza a las infecciones es un equilibrio dinámico entre el huésped y el
microorganismo, cada uno con sus mecanismos ofensivos y defensivos, cada uno capaz de adaptarse a las fases
de una batalla cambiante.
1. En el caso de una persona normal
a. La inoculación de un número relativamente enorme de gérmenes no virulentos no causa
enfermedad.
b. La inoculación de un pequeño número de gérmenes virulentos puede no causar enfermedad.
c. La inoculación de un número elevado de gérmenes virulentos en un lugar inadecuado (puerta de
entrada) no causa enfermedad.
d. La inoculación de un número elevado (mayor de la dosis infectante mínima) de gérmenes
virulentos en un lugar adecuado origina infección
2. En el caso de una persona con resistencia elevada, incluso cuando gran número de gérmenes virulentos
alcanza la puerta de entrada adecuada, puede no producir enfermedad.
3. En el caso de una persona con poca resistencia (local o general), incluso una dosis pequeña de gérmenes con
poca virulencia causará infección.
Lámina 36. INFECCION POR PAPILOMAVIRUS HUMANO
Más de 80 tipos de papilomavirus humano han sido identificados y más de 10 entidades clínicas benignas y
malignas relacionadas con infección por algunos de estos tipos son conocidas. Las entidades clínicas más
comunes asociadas a infección por papilomavirus humano son: verruga vulgar, epidermodisplasia verruciformis,
condiloma acuminado, papulosis bowenoide, hiperplasia epitelial focal y carcinoma epidermoide (cutáneo,
genital o esofágico). Una característica esencial de estas infecciones es que el virus tiene un tropismo peculiar
por las células de los epitelios escamosos, particularmente las células basales. Desde el punto de vista biológico
las infecciones por papilomavirus pueden dividirse en dos grandes grupos: aquellas donde existe replicación
activa de virus en los epitelios y aquella donde existe incorporación del genoma viral en las células, lo que
induce una transformación maligna. En casos donde existe replicación viral activa a nivel citoplasmático, las
infecciones suelen ser autolimitadas y comúnmente resultan en lesiones aplanadas o elevadas, que son llamadas
verrugas planas o condilomas.
En el cuello uterino las lesiones aplanadas pueden ser causadas por alguno de los 22 tipos de papilomavirus
humano que infectan el tracto anogenital. En las lesiones planas las células se caracterizan por tener vacuolas
Lámina 36. Papilomavirus
Lámina 37. Citomegalovirus
Lámina 38. Helicobacter pylori
Lámina 39. Tuberculosis
Lámina 40. Leishmaniasis

56
citoplasmáticas y anormalidades nucleares. Esas lesiones han sido designadas como atipia coilocítica o
coilocitosis. Cuando las lesiones son de alto grado, frecuentemente son aneuploides y pueden progresar a
carcinoma invasivo. En estos casos las lesiones están compuestas de células basales atípicas proliferantes con
una elevada relación núcleo/citoplasma. En contraste con las lesiones de bajo grado en que los tipos asociados de
papilomavirus humano son muy heterogéneos, las lesiones de alto grado suelen estar asociadas con un limitado
número de papilomavirus altamente oncogénicos, incluyendo HPV 16, 18 y 31. La prevalencia de papilomavirus
humano en ambos tipos de lesiones es similar, aproximadamente 90%. En lesiones de bajo grado existe
producción de partículas virales y en consecuencia el virus es contagioso. En lesiones de alto grado el ADN viral
está presente, pero partículas virales infecciosas son producidas raramente o en cantidades muy pequeñas.
Historia Clínica. Paciente mujer a quien durante una colposcopía se le observa lesiones blanquecinas en el
cuello uterino. Se le realiza un papanicolaou en el que se observa coilocitos. Se le toma una biopsia de cuello
uterino.
La figura de la izquierda muestra la imagen colposcópica de un cuello uterino normal y hacia la derecha se
observa la imagen colposcópica de una displasia de bajo grado asociada a papilomavirus humano.
Lámina. Se observa un epitelio exocervical de espesor aumentado, que destaca por la palidez de las células
escamosas de los estratos superiores. A mayor aumento se nota que las células tienen unos núcleos convolutos,
Esta es la imagen característica de los
coilocitos tal como se les observa en un
preparado citológico teñido con la
coloración de Papanicolaou.
Se trata de células epiteliales escamosas que
pueden ser uni o binucleadas, con núcleos
hipercromáticos y de contornos irregulares,
con un halo perinuclear de color pálido

57
de contornos irregulares, con una cromatina que se tiñe intensamente y citoplasmas amplios, con halo
transparente alrededor de los núcleos. Esta alteración es más visible en las células de la superficie epitelial en
comparación con las células del estrato basal.
Lámina 37. INFECCION POR CITOMEGALOVIRUS
El citomegalovirus (CMV) se encuentra de manera universal en todas las localizaciones geográficas y en todos
los grupos socioeconómicos, aunque está más extendido en países en desarrollo y en áreas con pobres
condiciones socioeconómicas. La mayoría de las personas sanas que son infectadas por citomegalovirus sufren
pocos síntomas (aunque una vez que una persona ha sido infectada el virus quedará latente en el de por vida),
excepto en casos en que el sistema inmune esté deprimido debido a medicación o enfermedad. La transmisión
del CMV ocurre de persona a persona. La infección requiere contacto cercano o íntimo. El virus puede ser
transmitido por vía sexual, por la leche materna, por órganos transplantados y raramente por transfusión de
sangre. La mayoría de las infecciones con citomegalovirus no son diagnosticadas porque el virus normalmente
produce pocos o ningún síntoma y tiende a reactivarse intermitentemente sin síntomas. No obstante, las personas
que han sido infectadas con citomegalovirus, desarrollan anticuerpos que persisten en el cuerpo. Existen tests de
laboratorio para detectar los anticuerpos contra citomegalovirus y además, el virus puede ser cultivado a partir de
la orina, muestras de tejido, etc. para detectar las infecciones activas. Se pueden hacer tests cualitativos y
cuantitativos, permitiendo a los médicos monitorizar la carga viral de los pacientes infectados por
citomegalovirus.
Historia Clínica. Paciente varón con SIDA, se presenta con fiebre, dificultad respiratoria, tos no productiva e
infiltrados difusos en la radiografía de tórax. El paciente fallece a los pocos días de su hospitalización y se le
realiza necropsia.
Lámina. A menor aumento se observa un parénquima pulmonar con una arquitectura básicamente conservada.
A mayor aumento se observa numerosas células, predominantemente macrófagos alveolares, neumocitos y
células endoteliales, que presentan tamaño aumentado, con inclusiones intranucleares e intracitoplasmáticas. Las
inclusiones intranucleares son centrales, oscuras o púrpuras y están rodeadas por un halo transparente. Las
inclusiones citoplasmáticas, que no se encuentran en todas la células infectadas, aparecen como gránulos
basofílicos gruesos.

58
Lámina 38. GASTRITIS POR HELICOBACTER PYLORI
En 1875, científicos alemanes descubrieron bacterias espirales en el epitelio del estómago humano. Estas
bacterias no podían ser cultivadas y por consiguiente este descubrimiento se olvidó en aquel momento. En 1892,
el investigador italiano Giulio Bizzozero describió una serie de bacterias espirales que vivían en el ambiente
ácido del estómago de perros.
El profesor Walery Jaworski de la Universidad de Jagiellonian en Cracovia investigó sedimentos de lavados
gástricos obtenidos de humanos en 1899. Además de unas bacterias alargadas, también encontró bacterias con
una característica forma espiral, a las cuales llamó Vibrio rugula. Este investigador fue el primero en sugerir la
participación de este microorganismo en enfermedades gástricas. Aunque este trabajo fue incluido en el "Manual
de Enfermedades Gástricas", no tuvo mucho impacto debido a que estaba escrito en polaco.
Esta bacteria fue redescubierta en 1979 por el patólogo australiano Robin Warren, quien en investigaciones
posteriores (a partir de 1981) junto a Barry Marshall, aisló este microorganismo de las mucosas de estómagos
humanos y fue el primero que consiguió cultivarla. En el trabajo original, Warren y Marshall afirmaron que
muchas de las úlceras estomacales y gastritis estaban causadas por la colonización del estómago por esta
bacteria, y no por estrés o comida picante como se sostenía hasta entonces.
Marshall y Warren posteriormente descubrieron que los antibióticos eran efectivos para el tratamiento de la
gastritis. En 1994, el National Institutes of Health reportó que las úlceras gástricas más comunes eran causadas
por H. pylori, y recomendaron el uso de antibióticos, siendo incluidos en el régimen de tratamiento. En el año
2005, Warren y Marshall fueron galardonados con el Premio Nobel de Medicina por sus trabajos acerca de H.
pylori.
Helicobacter pylori es una bacteria que infecta el mucus del epitelio estomacal humano. Muchas úlceras y
algunos tipos de gastritis son debidas a infecciones por H. pylori. En muchos casos, los sujetos infectados nunca
llegan a desarrollar ningún tipo de síntoma. Esta bacteria vive exclusivamente en el estómago humano, siendo el
único organismo conocido que puede subsistir en un ambiente tan ácido. Es una bacteria espiral (de esta
característica morfológica deriva el nombre de Helicobacter) y puede "atornillarse" literalmente por sí misma
para colonizar el epitelio estomacal.
Historia Clínica: Paciente mujer de 32 años que acude al servicio de Gastroenterología por dolor tipo ardor en
el epigastrio, de tres meses de evolución, que calma con el uso de antiácidos. En la endoscopía se encuentra una
mucosa gástrica eritematosa a nivel de la región antropilórica.

59
Lámina: Biopsia gástrica, región antral. Se observa que el infiltrado inflamatorio con algunos
polimorfonucleares invade las glándulas, lo que indica actividad del componente inflamatorio agudo. Cuando la
inflamación es de mayor duración se observa formación de acúmulos linfoides. Los estadios agudo y crónico
pueden coexistir en casos de inflamación crónica reagudizada. Los bacilos alargados correspondientes a
Helicobacter pylori se pueden encontrar en la superficie mucosa con las coloraciones convencionales, aunque
destacan más fácilmente con coloraciones basadas en plata como Warthin Starry.
Lámina 39. TUBERCULOSIS PULMONAR
Mycobacterium tuberculosis es un bacilo delgado, recto o ligeramente curvo, aerobio estricto, que crece mejor
en medios ligeramente ácidos. Un rasgo importante es su resistencia a la desecación y a los antisépticos usuales,
lo que facilita la diseminación de la infección. Otra característica significativa es su resistencia a la tinción por
métodos comunes y la fijación que se logra después que han tomado el colorante, pues no se decoloran con
alcohol ni ácido, debido a su gran contenido de lípidos. (Se les llama acido alcohol resistentes). Cuando una
persona sin exposición previa y con adecuada respuesta inmune inhala bacilos tuberculosos, estos se localizan en
la porción inferior de uno de los lóbulos pulmonares superiores, a 3 a 4 mm. de la superficie pleural, en un
conducto alveolar. A las pocas horas aparecen granulocitos que ingieren los bacilos, pero esta respuesta es débil
y en unas 24 horas los granulocitos mueren y liberan bacilos viables. En este momento macrófagos han llegado
al foco de inflamación y fagocitan los bacilos, algunas de estas células permanecen en el lugar de ataque donde
forman el complejo primario parenquimatoso y otros migran a los linfáticos regionales transportando bacilos
tuberculosos viables para formar focos linfógenos. Se necesitan unas dos semanas para que se acumule un
número suficiente de macrófagos (células epitelioides) para formar un tubérculo epitelioide diminuto, de un
milímetro de diámetro. Este continua creciendo y poco después aparecen en su centro una o varias células
gigantes de Langhans, de unas 200 a 300 micras de diámetro con numerosos núcleos, dispuestos
característicamente en forma de herradura, anillo o acúmulos, según el plano de corte del tejido.
Aproximadamente en este tiempo la parte central del tubérculo comienza a experimentar necrosis por
caseificación, fenómeno simultáneo con el desarrollo de la reacción de hipersensibilidad retardada que
caracteriza a esta enfermedad. Durante este tiempo continúa formándose una zona cada vez más gruesa de
células epitelioides alrededor del área central de necrosis caseosa. Al evolucionar la resistencia adquirida las
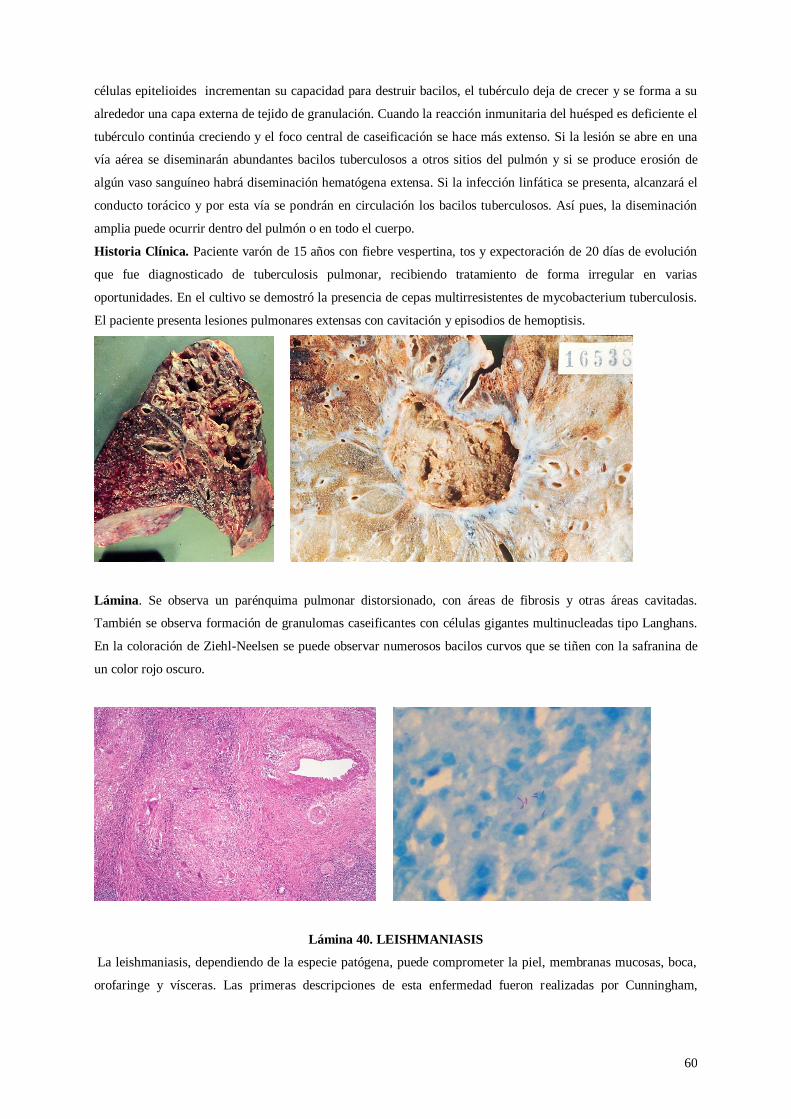
60
células epitelioides incrementan su capacidad para destruir bacilos, el tubérculo deja de crecer y se forma a su
alrededor una capa externa de tejido de granulación. Cuando la reacción inmunitaria del huésped es deficiente el
tubérculo continúa creciendo y el foco central de caseificación se hace más extenso. Si la lesión se abre en una
vía aérea se diseminarán abundantes bacilos tuberculosos a otros sitios del pulmón y si se produce erosión de
algún vaso sanguíneo habrá diseminación hematógena extensa. Si la infección linfática se presenta, alcanzará el
conducto torácico y por esta vía se pondrán en circulación los bacilos tuberculosos. Así pues, la diseminación
amplia puede ocurrir dentro del pulmón o en todo el cuerpo.
Historia Clínica. Paciente varón de 15 años con fiebre vespertina, tos y expectoración de 20 días de evolución
que fue diagnosticado de tuberculosis pulmonar, recibiendo tratamiento de forma irregular en varias
oportunidades. En el cultivo se demostró la presencia de cepas multirresistentes de mycobacterium tuberculosis.
El paciente presenta lesiones pulmonares extensas con cavitación y episodios de hemoptisis.
Lámina. Se observa un parénquima pulmonar distorsionado, con áreas de fibrosis y otras áreas cavitadas.
También se observa formación de granulomas caseificantes con células gigantes multinucleadas tipo Langhans.
En la coloración de Ziehl-Neelsen se puede observar numerosos bacilos curvos que se tiñen con la safranina de
un color rojo oscuro.
Lámina 40. LEISHMANIASIS
La leishmaniasis, dependiendo de la especie patógena, puede comprometer la piel, membranas mucosas, boca,
orofaringe y vísceras. Las primeras descripciones de esta enfermedad fueron realizadas por Cunningham,
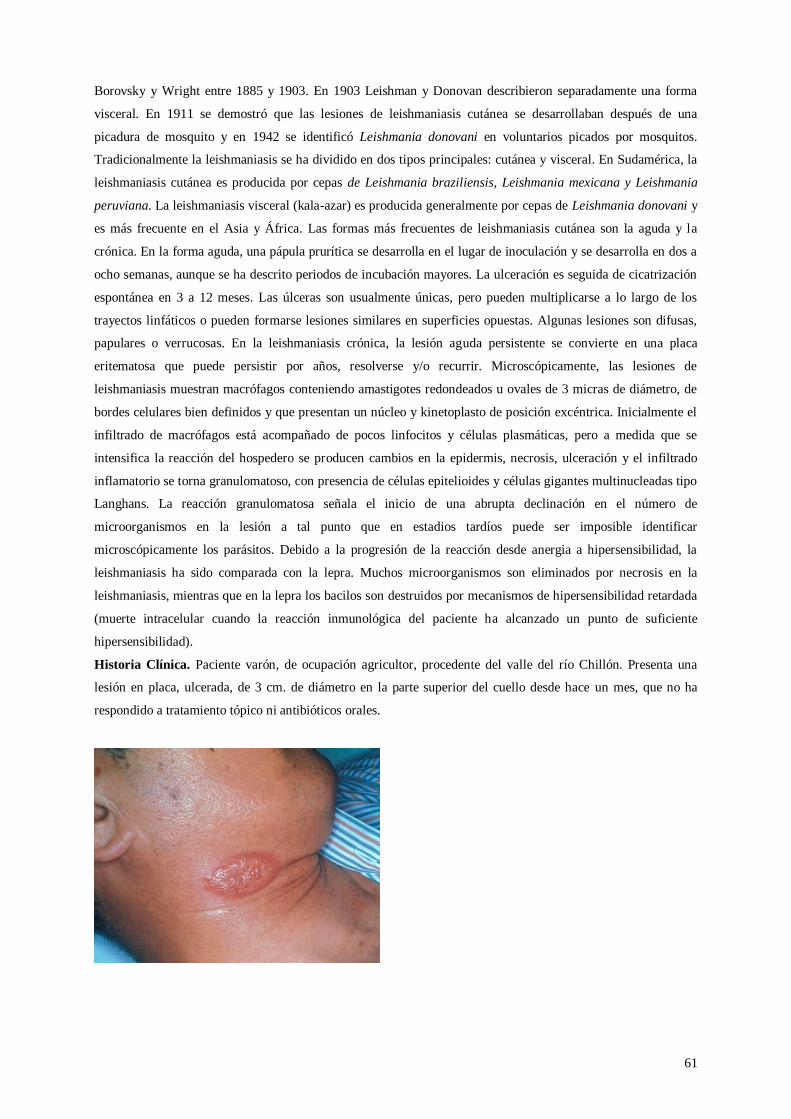
61
Borovsky y Wright entre 1885 y 1903. En 1903 Leishman y Donovan describieron separadamente una forma
visceral. En 1911 se demostró que las lesiones de leishmaniasis cutánea se desarrollaban después de una
picadura de mosquito y en 1942 se identificó Leishmania donovani en voluntarios picados por mosquitos.
Tradicionalmente la leishmaniasis se ha dividido en dos tipos principales: cutánea y visceral. En Sudamérica, la
leishmaniasis cutánea es producida por cepas de Leishmania braziliensis, Leishmania mexicana y Leishmania
peruviana. La leishmaniasis visceral (kala-azar) es producida generalmente por cepas de Leishmania donovani y
es más frecuente en el Asia y África. Las formas más frecuentes de leishmaniasis cutánea son la aguda y la
crónica. En la forma aguda, una pápula prurítica se desarrolla en el lugar de inoculación y se desarrolla en dos a
ocho semanas, aunque se ha descrito periodos de incubación mayores. La ulceración es seguida de cicatrización
espontánea en 3 a 12 meses. Las úlceras son usualmente únicas, pero pueden multiplicarse a lo largo de los
trayectos linfáticos o pueden formarse lesiones similares en superficies opuestas. Algunas lesiones son difusas,
papulares o verrucosas. En la leishmaniasis crónica, la lesión aguda persistente se convierte en una placa
eritematosa que puede persistir por años, resolverse y/o recurrir. Microscópicamente, las lesiones de
leishmaniasis muestran macrófagos conteniendo amastigotes redondeados u ovales de 3 micras de diámetro, de
bordes celulares bien definidos y que presentan un núcleo y kinetoplasto de posición excéntrica. Inicialmente el
infiltrado de macrófagos está acompañado de pocos linfocitos y células plasmáticas, pero a medida que se
intensifica la reacción del hospedero se producen cambios en la epidermis, necrosis, ulceración y el infiltrado
inflamatorio se torna granulomatoso, con presencia de células epitelioides y células gigantes multinucleadas tipo
Langhans. La reacción granulomatosa señala el inicio de una abrupta declinación en el número de
microorganismos en la lesión a tal punto que en estadios tardíos puede ser imposible identificar
microscópicamente los parásitos. Debido a la progresión de la reacción desde anergia a hipersensibilidad, la
leishmaniasis ha sido comparada con la lepra. Muchos microorganismos son eliminados por necrosis en la
leishmaniasis, mientras que en la lepra los bacilos son destruidos por mecanismos de hipersensibilidad retardada
(muerte intracelular cuando la reacción inmunológica del paciente ha alcanzado un punto de suficiente
hipersensibilidad).
Historia Clínica. Paciente varón, de ocupación agricultor, procedente del valle del río Chillón. Presenta una
lesión en placa, ulcerada, de 3 cm. de diámetro en la parte superior del cuello desde hace un mes, que no ha
respondido a tratamiento tópico ni antibióticos orales.
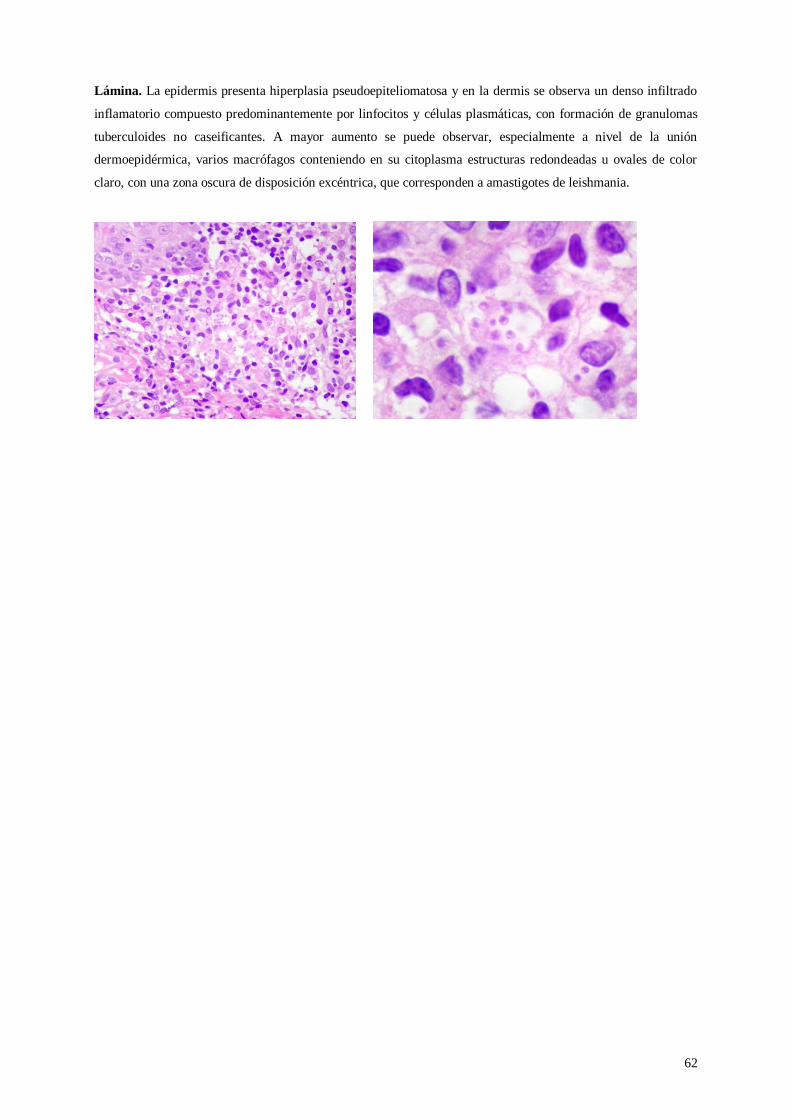
62
Lámina. La epidermis presenta hiperplasia pseudoepiteliomatosa y en la dermis se observa un denso infiltrado
inflamatorio compuesto predominantemente por linfocitos y células plasmáticas, con formación de granulomas
tuberculoides no caseificantes. A mayor aumento se puede observar, especialmente a nivel de la unión
dermoepidérmica, varios macrófagos conteniendo en su citoplasma estructuras redondeadas u ovales de color
claro, con una zona oscura de disposición excéntrica, que corresponden a amastigotes de leishmania.

63
PRÁCTICA Nº 9. ENFERMEDADES AMBIENTALES Y NUTRICIONALES
LÁMINA 41. MESOTELIOMA PULMONAR
El mesotelioma es un tumor derivado de las células mesoteliales que recubren la cavidad pleural, peritoneal o la
pericárdica, aunque es mucho más frecuente en la pleura que en las otras dos localizaciones. Casi siempre los
mesoteliomas pleurales están asociados con exposición ambiental a asbestos, que usualmente son utilizados en la
industria, por lo que se le considera una enfermedad ocupacional.
El mesotelioma se caracteriza por presentarse como un engrosamiento homogéneo y difuso de la pleura que se
continúa a nivel de las incisuras que dividen los lóbulos y segmentos pulmonares. Esto produce una imagen
característica en las radiografías o tomografías de tórax lo que facilita su diagnóstico. Es usual que los pacientes
se presenten con derrame pleural ipsilateral al lado afectado. Histológicamente el mesotelioma se caracteriza por
presentar una variedad de patrones morfológicos siendo uno de los más frecuentemente observados el llamado
mesotelioma fibroso.
Historia Clínica. Paciente varón de 56 años de edad que ha trabajado durante muchos años como obrero en una
empresa metal mecánica. Se presenta con dificultad respiratoria y dolor pleurítico en el hemitórax derecho. Al
examen se le encuentra signos de derrame pleural (matidez a la percusión del hemitórax inferior derecho y
ausencia de murmullo vesicular en esa zona). Una radiografía de tórax post drenaje del líquido pleural muestra
un engrosamiento homogéneo de la pleura, que se extiende siguiendo los espacios interlobulares. La tomografía
computarizada muestra una masa pleural lobular que envuelve el pulmón derecho.
Lámina. Se observa una proliferación fusocelular de patrón fibrosante constituída por células neoplásicas
Lámina 41. Mesotelioma pulmonar
Lámina 42. Hepatitis alcohólica
Lámina 43. Anemia aplásica
Lámina 44. Neumoconiosis
Lámina 45. Queratosis actínica

64
alargadas y dispersas en una matriz de tejido conectivo denso y en otra sección el patrón epitelioide que esta
neoplasia puede presentar de manera simultánea.
.
LÁMINA 42. HEPATITIS ALCOHÓLICA
El consumo excesivo de alcohol es la principal causa de enfermedad hepática en la mayor parte de países
occidentalizados. La ingestión aguda de hasta 80 gramos de alcohol generalmente produce cambios hepáticos
reversibles como el hígado graso. La ingesta diaria de al menos 80 gramos de alcohol genera un riesgo severo de
daño hepático y la ingestión diaria de al menos 160 gramos por 10 a 20 años está a menudo asociada con severo
daño hepático crónico. No obstante, sólo 10 a 15% de alcohólicos desarrollan cirrosis, algunas veces sin
antecedentes clínicos de esteatosis o hepatitis alcohólica.
La ingestión aguda de alcohol produce esteatosis microvesicular, independientemente del patrón individual de
consumo de alcohol. En contraste, el consumo crónico de alcohol tiene una variedad de efectos adversos e
induce tres formas diferentes de enfermedad hepática: esteatosis, hepatitis y cirrosis. Estas tres formas de
enfermedad hepática son colectivamente llamadas enfermedad hepática alcohólica. Debido a que la esteatosis y
la hepatitis pueden desarrollarse independientemente una de otra, este patrón de enfermedad no necesariamente
representa un patrón progresivo de lesión hepática.
La hepatitis alcohólica es una enfermedad aguda que usualmente es consecuencia del consumo agudo de alcohol.
Los síntomas y anormalidades de laboratorio de esta enfermedad varían desde mínimos hasta severos (falla
hepática fulminante). Entre esos extremos los pacientes pueden presentar síntomas inespecíficos como malestar
general, anorexia, pérdida de peso, malestar en hemiabdomen inferior y hepatomegalia dolorosa. Los exámenes
de laboratorio generalmente indican hiperbilirrubinemia, niveles elevados de fosfatasa alcalina y leucocitosis con
neutrofilia. Con episodios repetidos de hepatitis alcohólica un tercio de pacientes desarrollan cirrosis al cabo de
unos años. La hepatitis alcohólica suele revertirse con una adecuada alimentación y el cese del consumo de
alcohol; no obstante, en algunos pacientes la hepatitis persiste a pesar de la abstinencia y eventualmente progresa
a cirrosis.
Historia Clínica. Paciente varón de 45 años con historia de abuso crónico de alcohol. Es evaluado por presentar,
después de una ingesta aguda de alcohol, ictericia (coloración amarilla de piel y escleras) y dolor abdominal. En
sus exámenes de laboratorio se observa hiperbilirrubinemia a predominio directa con aumento de niveles de
transaminasas.
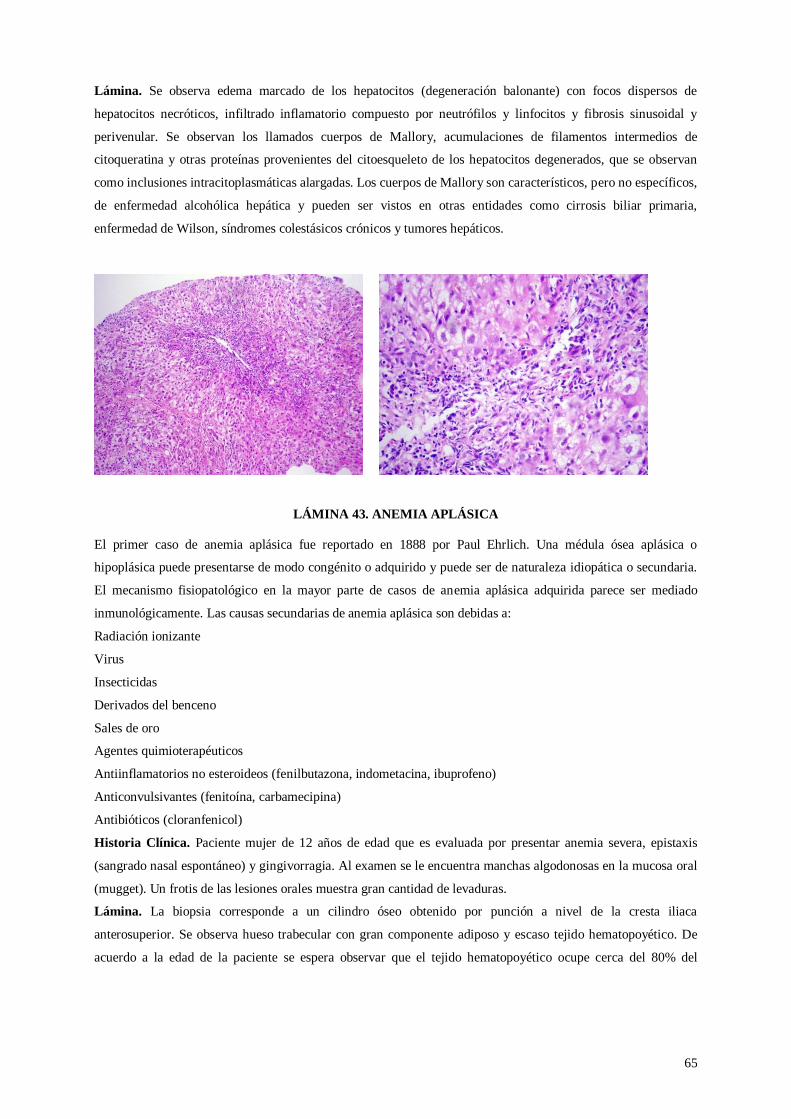
65
Lámina. Se observa edema marcado de los hepatocitos (degeneración balonante) con focos dispersos de
hepatocitos necróticos, infiltrado inflamatorio compuesto por neutrófilos y linfocitos y fibrosis sinusoidal y
perivenular. Se observan los llamados cuerpos de Mallory, acumulaciones de filamentos intermedios de
citoqueratina y otras proteínas provenientes del citoesqueleto de los hepatocitos degenerados, que se observan
como inclusiones intracitoplasmáticas alargadas. Los cuerpos de Mallory son característicos, pero no específicos,
de enfermedad alcohólica hepática y pueden ser vistos en otras entidades como cirrosis biliar primaria,
enfermedad de Wilson, síndromes colestásicos crónicos y tumores hepáticos.
LÁMINA 43. ANEMIA APLÁSICA
El primer caso de anemia aplásica fue reportado en 1888 por Paul Ehrlich. Una médula ósea aplásica o
hipoplásica puede presentarse de modo congénito o adquirido y puede ser de naturaleza idiopática o secundaria.
El mecanismo fisiopatológico en la mayor parte de casos de anemia aplásica adquirida parece ser mediado
inmunológicamente. Las causas secundarias de anemia aplásica son debidas a:
Radiación ionizante
Virus
Insecticidas
Derivados del benceno
Sales de oro
Agentes quimioterapéuticos
Antiinflamatorios no esteroideos (fenilbutazona, indometacina, ibuprofeno)
Anticonvulsivantes (fenitoína, carbamecipina)
Antibióticos (cloranfenicol)
Historia Clínica. Paciente mujer de 12 años de edad que es evaluada por presentar anemia severa, epistaxis
(sangrado nasal espontáneo) y gingivorragia. Al examen se le encuentra manchas algodonosas en la mucosa oral
(mugget). Un frotis de las lesiones orales muestra gran cantidad de levaduras.
Lámina. La biopsia corresponde a un cilindro óseo obtenido por punción a nivel de la cresta iliaca
anterosuperior. Se observa hueso trabecular con gran componente adiposo y escaso tejido hematopoyético. De
acuerdo a la edad de la paciente se espera observar que el tejido hematopoyético ocupe cerca del 80% del
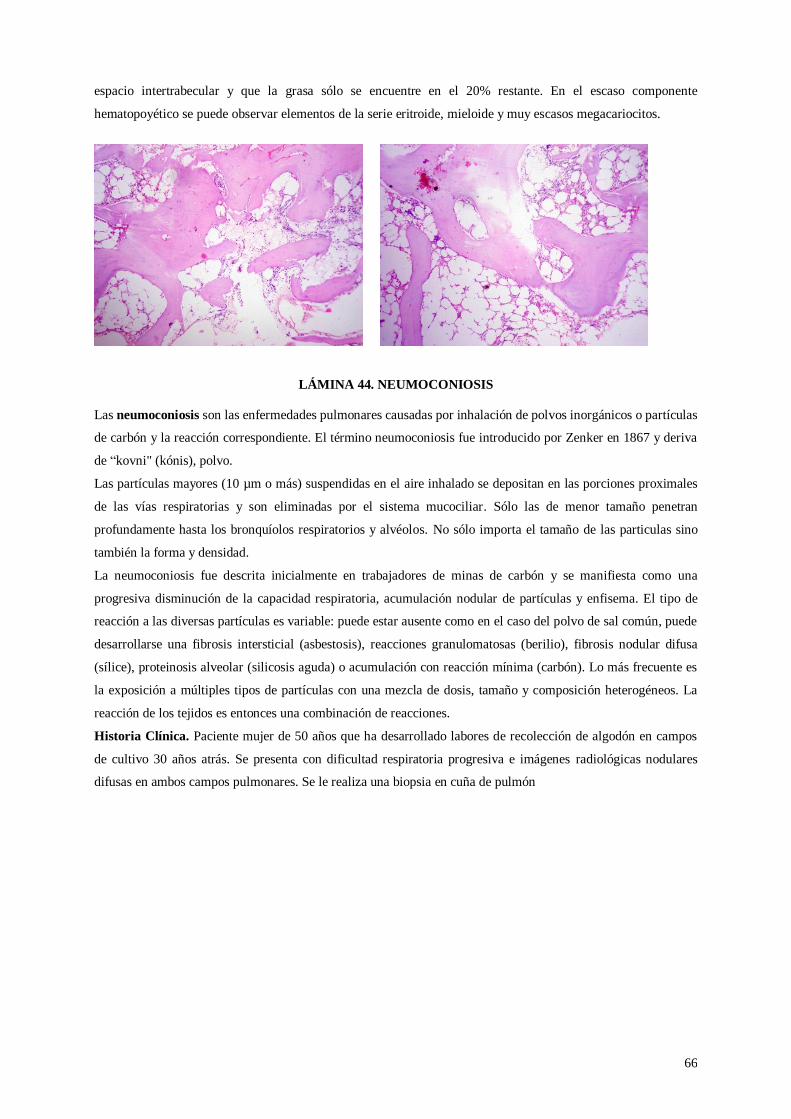
66
espacio intertrabecular y que la grasa sólo se encuentre en el 20% restante. En el escaso componente
hematopoyético se puede observar elementos de la serie eritroide, mieloide y muy escasos megacariocitos.
LÁMINA 44. NEUMOCONIOSIS
Las neumoconiosis son las enfermedades pulmonares causadas por inhalación de polvos inorgánicos o partículas
de carbón y la reacción correspondiente. El término neumoconiosis fue introducido por Zenker en 1867 y deriva
de “kovni" (kónis), polvo.
Las partículas mayores (10 µm o más) suspendidas en el aire inhalado se depositan en las porciones proximales
de las vías respiratorias y son eliminadas por el sistema mucociliar. Sólo las de menor tamaño penetran
profundamente hasta los bronquíolos respiratorios y alvéolos. No sólo importa el tamaño de las particulas sino
también la forma y densidad.
La neumoconiosis fue descrita inicialmente en trabajadores de minas de carbón y se manifiesta como una
progresiva disminución de la capacidad respiratoria, acumulación nodular de partículas y enfisema. El tipo de
reacción a las diversas partículas es variable: puede estar ausente como en el caso del polvo de sal común, puede
desarrollarse una fibrosis intersticial (asbestosis), reacciones granulomatosas (berilio), fibrosis nodular difusa
(sílice), proteinosis alveolar (silicosis aguda) o acumulación con reacción mínima (carbón). Lo más frecuente es
la exposición a múltiples tipos de partículas con una mezcla de dosis, tamaño y composición heterogéneos. La
reacción de los tejidos es entonces una combinación de reacciones.
Historia Clínica. Paciente mujer de 50 años que ha desarrollado labores de recolección de algodón en campos
de cultivo 30 años atrás. Se presenta con dificultad respiratoria progresiva e imágenes radiológicas nodulares
difusas en ambos campos pulmonares. Se le realiza una biopsia en cuña de pulmón

67
Lámina. Se observa lesiones parenquimales nodulares dispersas. En cada una de estas lesiones se observa un
infiltrado granulomatoso compuesto principalmente por macrófagos, células gigantes multinucleadas a cuerpo
extraño y también por linfocitos. Llama la atención la presencia de estructuras amorfas refringentes de naturaleza
vegetal en relación con los nódulos descritos.
LÁMINA 45. QUERATOSIS ACTÍNICA
Existe una variedad de lesiones cutáneas, preneoplásicas, neoplásicas y no neoplásicas, relacionadas con
exposición solar. Las neoplasias cutáneas usualmente se originan en células de la epidermis. La excepción a esta
regla es el fibroxantoma atípico, una neoplasia maligna muy poco frecuente que se origina en la dermis. La
lesión cutánea preneoplásica característicamente asociada a exposición solar es la queratosis actínica. Las
neoplasias asociadas a exposición solar que se originan en las células de la epidermis son algunas formas de
carcinoma epidermoide, el carcinoma basocelular y el melanoma.
La queratosis actínica, también llamada queratosis solar, es una lesión frecuente que se presenta en personas
adultas o adultos mayores en relación directa con radiación UVB. Clínicamente se presentan como lesiones
hiperqueratósicas que pueden ser hiperpigmentadas, localizadas en cara, cuello, dorso de manos y antebrazos. El
examen histológico es necesario para establecer el diagnóstico porque las lesiones clínicas pueden ser
confundidas con nevus, melanoma y carcinoma basocelular principalmente. El estudio genético de los
queratinocitos displásicos de la queratosis actínica muestra múltiples aneuploidías y frecuente mutación del gen
p53. Cerca del 80% de carcinomas epidermoides que se originan en zonas fotoexpuestas están asociados con
queratosis actínicas como lesiones precursoras.
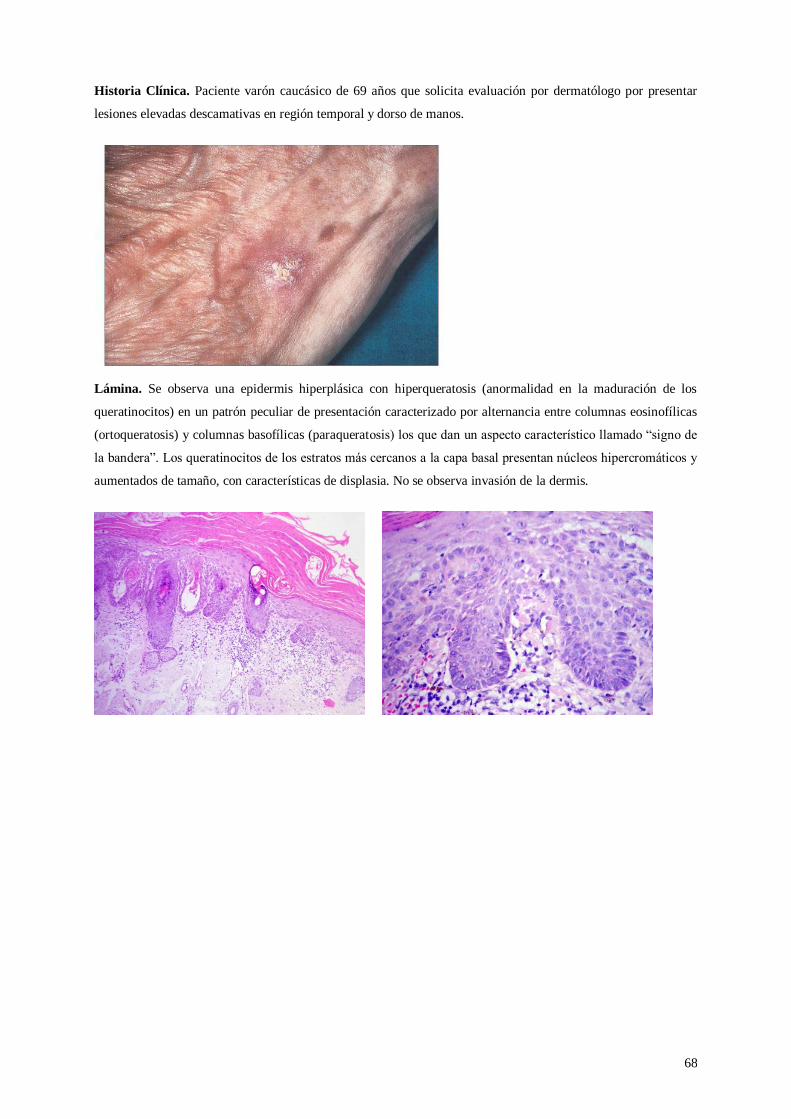
68
Historia Clínica. Paciente varón caucásico de 69 años que solicita evaluación por dermatólogo por presentar
lesiones elevadas descamativas en región temporal y dorso de manos.
Lámina. Se observa una epidermis hiperplásica con hiperqueratosis (anormalidad en la maduración de los
queratinocitos) en un patrón peculiar de presentación caracterizado por alternancia entre columnas eosinofílicas
(ortoqueratosis) y columnas basofílicas (paraqueratosis) los que dan un aspecto característico llamado “signo de
la bandera”. Los queratinocitos de los estratos más cercanos a la capa basal presentan núcleos hipercromáticos y
aumentados de tamaño, con características de displasia. No se observa invasión de la dermis.

69
PRÁCTICA N° 10: INMUNOPATOLOGÍA
La respuesta inmunitaria es un mecanismo de defensa normal frente a factores ambientales adversos como
microorganismos y tóxicos; por lo general es eficaz pero pueden producirse enfermedades por:
Una respuesta inmunitaria insuficiente
Una respuesta inmunitaria excesiva
Una respuesta inmunitaria no deseada o inadecuada: puede producir enfermedades autoinmunes.
Lámina 46. PÉNFIGO VULGAR
En la piel se pueden formar vesículas y ampollas por diferentes causas, mecanismos y a diferentes niveles de la
epidermis, incluyendo la unión dermoepidérmica. El pénfigo pertenece al grupo de enfermedades ampollares
autoinmunes, bastante raras, caracterizadas por ampollas y erosiones de la piel y de las membranas mucosas. Se
clasifica en cuatro tipos: pénfigo vulgar, pénfigo foliáceo, pénfigo paraneoplásico y pénfigo por IgA. El pénfigo
vulgar es una enfermedad dermatológica sistémica grave que afecta las membranas mucosas de la boca y la piel
y que se manifiesta como ampollas grandes y frágiles llenas de un fluído claro. En esta enfermedad existen
anticuerpos dirigidos contra el llamado “antígeno del pénfigo vulgar”, también llamado desmogleína 3, un
polipéptido que es miembro de la subfamilia de las desmogleínas y de la familia de las cadherinas. Este antígeno
está localizado en los desmosomas de los queratinocitos. Más del 50% de pacientes con pénfigo vulgar tienen
también anticuerpos contra desmogleína 1, que también tienen importancia patogénica. Los anticuerpos al unirse
a los antígenos producen separación (acantolisis) de los queratinocitos. En el pénfigo vulgar los anticuerpos
producidos por los linfocitos B circulantes tienden a unirse preferentemente a desmosomas de la epidermis
inferior, lo que refleja diferencias en la expresión relativa de diferentes cadherinas a diferentes niveles de la
epidermis.
Historia clínica: Varón de 27 años de edad que hace 2 semanas presenta bruscamente lesiones ampollares
distribuidas en todo el cuerpo. Las lesiones evolucionan hasta convertirse en costras, algunas con exudado.
Lámina 46. Pénfigo vulgar
Lámina 47. Glomerulonefritis lúpica
Lámina 48. Lepra lepromatosa
Lámina 49. Poliarteritis nodosa
Lámina 50. Nódulo reumatoide
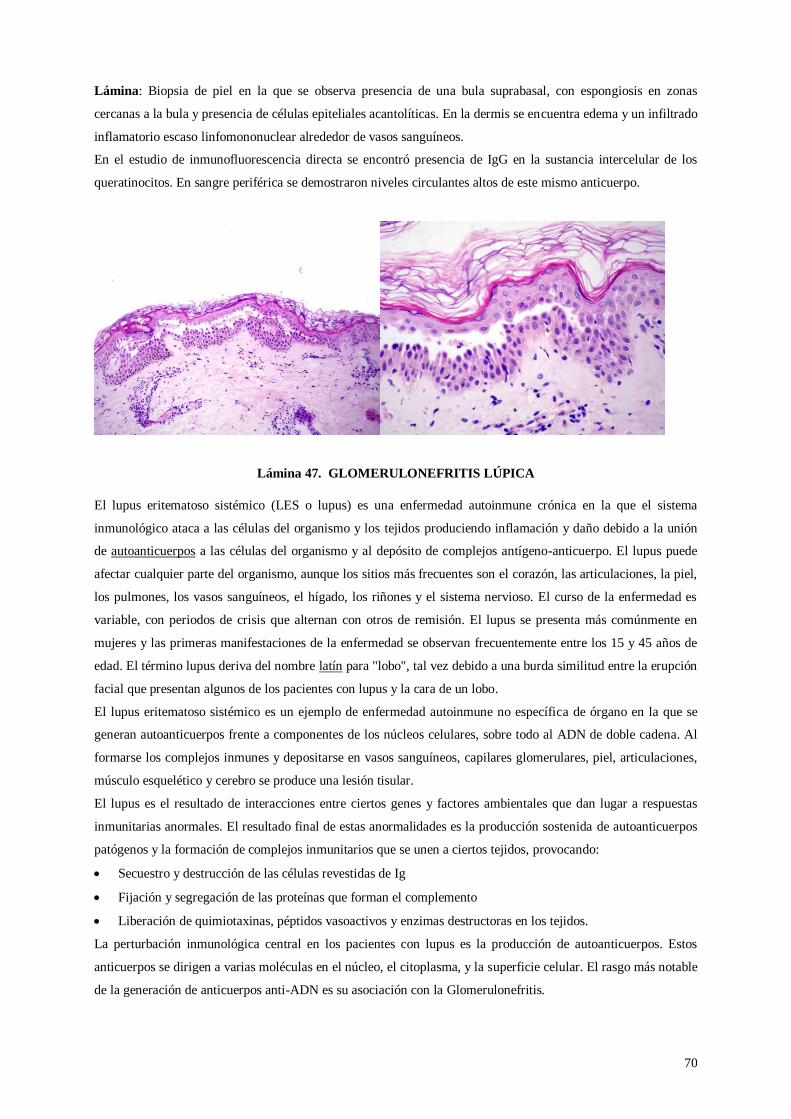
70
Lámina: Biopsia de piel en la que se observa presencia de una bula suprabasal, con espongiosis en zonas
cercanas a la bula y presencia de células epiteliales acantolíticas. En la dermis se encuentra edema y un infiltrado
inflamatorio escaso linfomononuclear alrededor de vasos sanguíneos.
En el estudio de inmunofluorescencia directa se encontró presencia de IgG en la sustancia intercelular de los
queratinocitos. En sangre periférica se demostraron niveles circulantes altos de este mismo anticuerpo.
Lámina 47. GLOMERULONEFRITIS LÚPICA
El lupus eritematoso sistémico (LES o lupus) es una enfermedad autoinmune crónica en la que el sistema
inmunológico ataca a las células del organismo y los tejidos produciendo inflamación y daño debido a la unión
de autoanticuerpos a las células del organismo y al depósito de complejos antígeno-anticuerpo. El lupus puede
afectar cualquier parte del organismo, aunque los sitios más frecuentes son el corazón, las articulaciones, la piel,
los pulmones, los vasos sanguíneos, el hígado, los riñones y el sistema nervioso. El curso de la enfermedad es
variable, con periodos de crisis que alternan con otros de remisión. El lupus se presenta más comúnmente en
mujeres y las primeras manifestaciones de la enfermedad se observan frecuentemente entre los 15 y 45 años de
edad. El término lupus deriva del nombre latín para "lobo", tal vez debido a una burda similitud entre la erupción
facial que presentan algunos de los pacientes con lupus y la cara de un lobo.
El lupus eritematoso sistémico es un ejemplo de enfermedad autoinmune no específica de órgano en la que se
generan autoanticuerpos frente a componentes de los núcleos celulares, sobre todo al ADN de doble cadena. Al
formarse los complejos inmunes y depositarse en vasos sanguíneos, capilares glomerulares, piel, articulaciones,
músculo esquelético y cerebro se produce una lesión tisular.
El lupus es el resultado de interacciones entre ciertos genes y factores ambientales que dan lugar a respuestas
inmunitarias anormales. El resultado final de estas anormalidades es la producción sostenida de autoanticuerpos
patógenos y la formación de complejos inmunitarios que se unen a ciertos tejidos, provocando:
Secuestro y destrucción de las células revestidas de Ig
Fijación y segregación de las proteínas que forman el complemento
Liberación de quimiotaxinas, péptidos vasoactivos y enzimas destructoras en los tejidos.
La perturbación inmunológica central en los pacientes con lupus es la producción de autoanticuerpos. Estos
anticuerpos se dirigen a varias moléculas en el núcleo, el citoplasma, y la superficie celular. El rasgo más notable
de la generación de anticuerpos anti-ADN es su asociación con la Glomerulonefritis.

71
Historia clínica: Paciente mujer de 25 años de edad con diagnóstico de lupus eritematoso sistémico hace cuatro
años. Actualmente presenta insuficiencia renal.
Exámenes auxiliares: Creatinina 2.5 mg/dl (V.N: < 1.4 mg/dL), úrea 60mg/dl (V.N: 10-40 mg/dL). Sedimento
urinario: cilindros leucocitarios, leucocitos 50 x campo. Urocultivo: negativo.
Lámina: Biopsia renal. En los glomérulos se observan zonas con engrosamiento de la membrana basal,
hiperplasia de células mesangiales, infiltrado de polimorfonucleares, cariorrexis o fragmentación nuclear, "asas
de alambre", crecientes o medialunas fibróticas y focos de necrosis fibrinoide.
Lámina 48. LEPRA LEPROMATOSA
Dos años antes que Roberto Koch descubriera el germen que causa la tuberculosis, Hansen (1878) descubrió el
Mycobacterium leprae agente causal de la lepra, una enfermedad causada por bacilos acido alcohol resistentes.
La lepra tuberculoide sigue las leyes de Lewandowsky y exhibe una fuerte reacción tisular epitelioide en la que
escasos o ningún bacilo puede ser demostrado. La imagen histológica de esta forma de lepra puede ser muy
parecida a sarcoidosis, lo que dificulta su diagnóstico. Por otro lado, en la lepra lepromatosa se encuentra
numerosos bacilos y la reacción tisular es menor, aunque muestra imágenes muy características en la que
predominan acumulaciones nodulares de histiocitos. Estos histiocitos, también llamados células de lepra o
células de Virchow, no tienen el aspecto de células epitelioides, sino más bien de células espumosas debido a su
alto contenido de bacilos y lípidos no polarizantes y deben ser considerados un tipo especial de reacción a cuerpo
extraño más que una expresión de inmunidad. Desde el punto de vista patológico, la lepra se divide en dos
formas polares: la lepra lepromatosa y la lepra tuberculoide, cada una con formas intermedias. Histológicamente
es más fácil comprender los hallazgos recordando en primer lugar que los bacilos primariamente comprometen
nervios y, dentro de los nervios, a las células de Schwann, las que los fagocitan y almacenan. El resto del cuadro
histológico depende de la capacidad del hospedero para reaccionar contra esto. Si la inmunidad celular
permanece baja y no se desarrolla una reacción de Mitsuda positiva, los macrófagos tisulares fagocitan los
bacilos y se llenan con acúmulos de éstos. De otro lado, si se desarrolla una reacción de inmunidad tisular, los
histiocitos destruyen a los bacilos y forman granulomas tuberculoides. Estados reaccionales parecen estar
relacionados con redireccionamientos de la inmunidad, a veces durante el curso del tratamiento y se presentan
como agudizaciones de la enfermedad. La exacerbación del proceso patológico es la llamada “reacción leproide”
en la lepra lepromatosa. Otra manifestación es el eritema nodoso leproso, que es la producción de lesiones

72
nodulares dolorosas en las que se ven numerosos bacilos en proceso de degeneración. El fenómeno de Lucio
representa una vasculitis necrotizante con muchos bacilos en casos de lepra difusa en la que toda la piel es
afectada y que se manifiesta como úlceras superficiales. La lepra histioide ha sido descrita como numerosas
colecciones de histiocitos separados por densas fibras de colágeno en la dermis profunda. En este caso las
lesiones clínicas son nódulos protuberantes.
Historia clínica: Paciente mujer de 28 años de edad, procedente de Madre de Dios, con historia de tres años de
lesiones dérmicas simétricas maculares en todo el cuerpo. Presenta pérdida de cejas y al examen se evidencia
polineuropatía sensorial. Test cutáneo de lepromina (Mitsuda): negativo. Anticuerpos anti M. Leprae: niveles
altos.
Lámina: Biopsia de piel. Se observa una epidermis adelgazada y en toda la dermis presencia de macrófagos
pobremente organizados que comprometen filetes nerviosos. La mayoría de macrófagos tiene un citoplasma
espumoso. Dentro del infiltrado el número de linfocitos es escaso.
Lámina Nº 49. POLIARTERITIS NODOSA
Esta enfermedad fue descrita originalmente por Kussmaul y Maier en 1866. La poliarteritis nodosa es una
enfermedad multisistémica que se presenta con una gran variedad de signos y síntomas. Se considera que es una
vasculitis predominantemente (pero no exclusivamente) de vasos de mediano calibre. Afecta más a varones que
a mujeres y ocurre a cualquier edad, mayoritariamente entre los 40 a 60 años. Los pacientes usualmente se
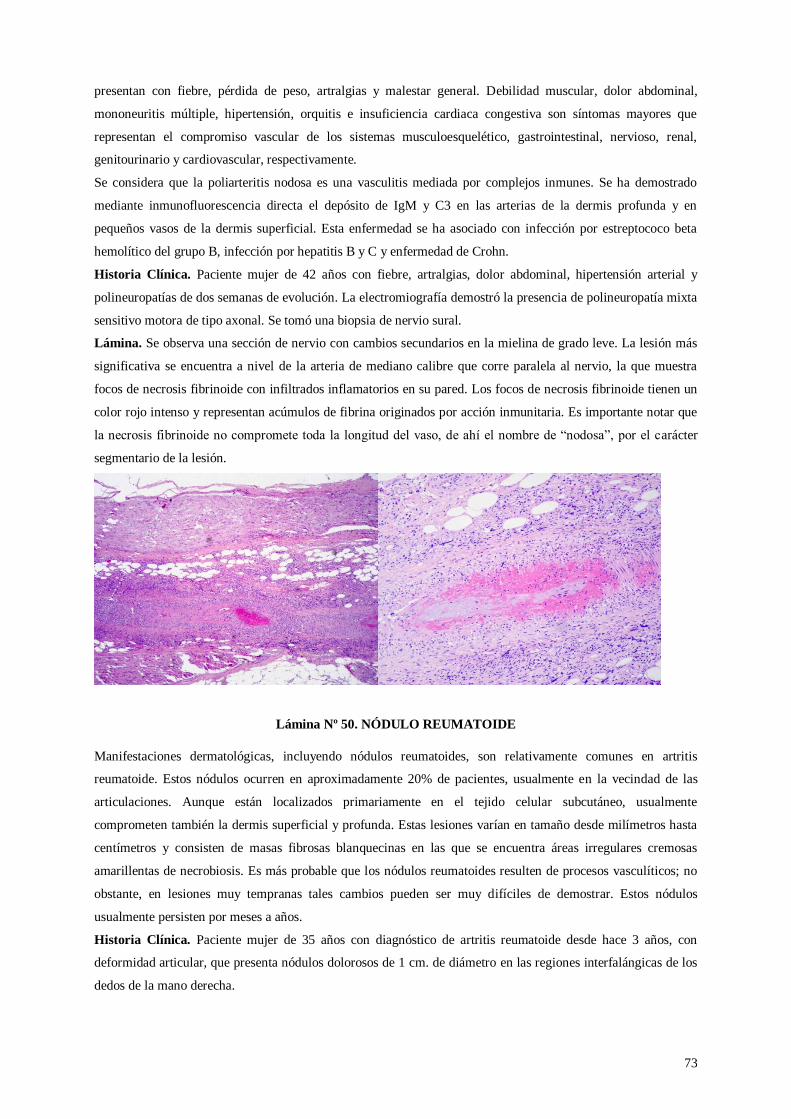
73
presentan con fiebre, pérdida de peso, artralgias y malestar general. Debilidad muscular, dolor abdominal,
mononeuritis múltiple, hipertensión, orquitis e insuficiencia cardiaca congestiva son síntomas mayores que
representan el compromiso vascular de los sistemas musculoesquelético, gastrointestinal, nervioso, renal,
genitourinario y cardiovascular, respectivamente.
Se considera que la poliarteritis nodosa es una vasculitis mediada por complejos inmunes. Se ha demostrado
mediante inmunofluorescencia directa el depósito de IgM y C3 en las arterias de la dermis profunda y en
pequeños vasos de la dermis superficial. Esta enfermedad se ha asociado con infección por estreptococo beta
hemolítico del grupo B, infección por hepatitis B y C y enfermedad de Crohn.
Historia Clínica. Paciente mujer de 42 años con fiebre, artralgias, dolor abdominal, hipertensión arterial y
polineuropatías de dos semanas de evolución. La electromiografía demostró la presencia de polineuropatía mixta
sensitivo motora de tipo axonal. Se tomó una biopsia de nervio sural.
Lámina. Se observa una sección de nervio con cambios secundarios en la mielina de grado leve. La lesión más
significativa se encuentra a nivel de la arteria de mediano calibre que corre paralela al nervio, la que muestra
focos de necrosis fibrinoide con infiltrados inflamatorios en su pared. Los focos de necrosis fibrinoide tienen un
color rojo intenso y representan acúmulos de fibrina originados por acción inmunitaria. Es importante notar que
la necrosis fibrinoide no compromete toda la longitud del vaso, de ahí el nombre de “nodosa”, por el carácter
segmentario de la lesión.
Lámina Nº 50. NÓDULO REUMATOIDE
Manifestaciones dermatológicas, incluyendo nódulos reumatoides, son relativamente comunes en artritis
reumatoide. Estos nódulos ocurren en aproximadamente 20% de pacientes, usualmente en la vecindad de las
articulaciones. Aunque están localizados primariamente en el tejido celular subcutáneo, usualmente
comprometen también la dermis superficial y profunda. Estas lesiones varían en tamaño desde milímetros hasta
centímetros y consisten de masas fibrosas blanquecinas en las que se encuentra áreas irregulares cremosas
amarillentas de necrobiosis. Es más probable que los nódulos reumatoides resulten de procesos vasculíticos; no
obstante, en lesiones muy tempranas tales cambios pueden ser muy difíciles de demostrar. Estos nódulos
usualmente persisten por meses a años.
Historia Clínica. Paciente mujer de 35 años con diagnóstico de artritis reumatoide desde hace 3 años, con
deformidad articular, que presenta nódulos dolorosos de 1 cm. de diámetro en las regiones interfalángicas de los
dedos de la mano derecha.
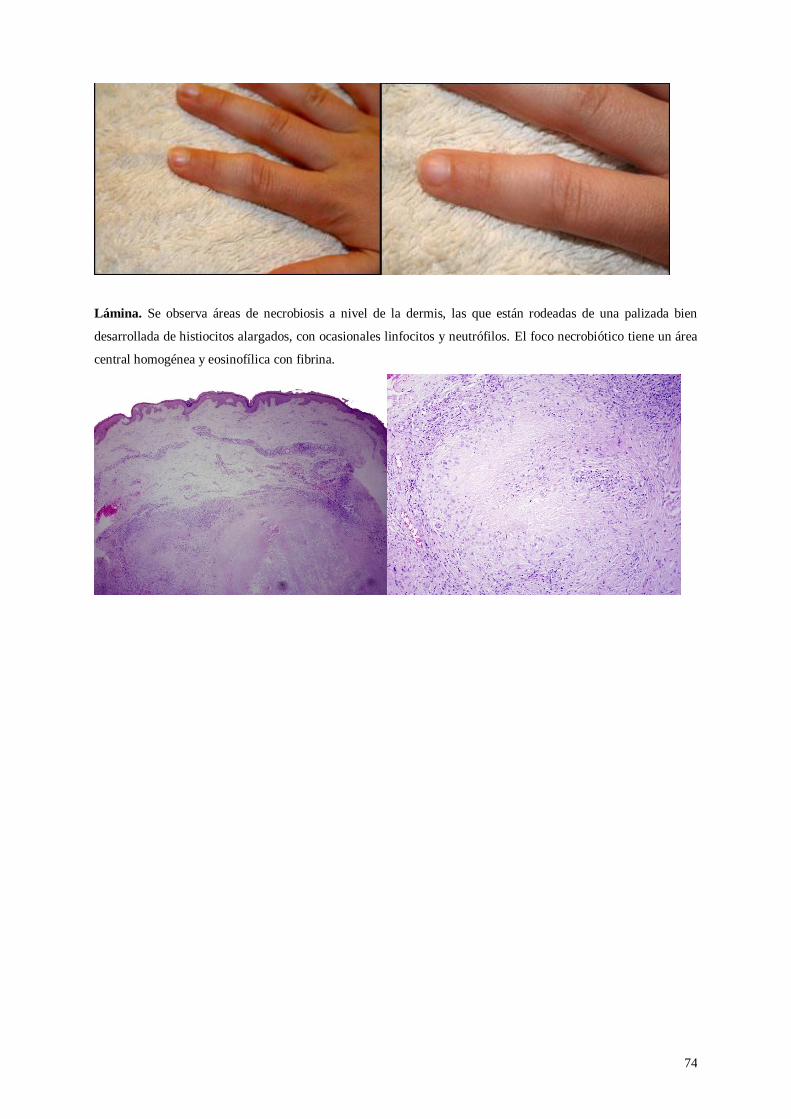
74
Lámina. Se observa áreas de necrobiosis a nivel de la dermis, las que están rodeadas de una palizada bien
desarrollada de histiocitos alargados, con ocasionales linfocitos y neutrófilos. El foco necrobiótico tiene un área
central homogénea y eosinofílica con fibrina.