
MANUAL Código: M-05-001-EQ - Promedan IPS
30
MANUAL Código: M-05-001-EQ TECNOVIGILANCIA Versión: 02 Vigencia: Septiembre de 2018 1. JUSTIFICACION Los dispositivos médicos son sometidos a diferentes controles durante su desarrollo, con el fin de garantizar la seguridad y efectividad durante su uso. No obstante, estos controles no son suficientes para garantizar que en la etapa pos-mercado no se presenten problemas o incidentes que puedan desencadenar daños para quienes los utilizan. Por este motivo, existe un consenso mundial sobre la necesidad de sistemas de regulación y vigilancia sobre todo el ciclo de vida de estas tecnologías. Para el caso de Colombia, el país cuenta con una regulación actualizada donde el Ministerio de Salud y Protección Social, el INVIMA, las Entidades Territoriales de Salud, los fabricantes e importadores de dispositivos médicos, tienen la responsabilidad de garantizar que este tipo de tecnologías cumplan con las condiciones clínicas de seguridad, eficacia y desempeño. El INVIMA, en su labor de inspección, vigilancia y control sobre los productos de su competencia, y en cumplimiento de las funciones descritas por la normatividad sanitaria vigente, lidera desde el 2008 el Programa Nacional de Tecnovigilancia. El objetivo principal de este programa es mejorar la protección de la salud y la seguridad de pacientes, usuarios y otros actores involucrados, mediante el control y reducción del riesgo asociado al uso de los dispositivos médicos comercializados en el territorio colombiano. En este sentido, el Instituto ha establecido los mecanismos para recolectar, evaluar y gestionar la información relacionada con la seguridad de los dispositivos médicos, con el propósito de tomar las medidas a que haya lugar para el cumplimiento de este objetivo. Es responsabilidad social y ética de todas las personas involucradas en la fabricación, comercialización, distribución, prescripción, manipulación y uso de los dispositivos médicos informar a la autoridad sanitaria cuando se tenga conocimiento sobre algún evento o incidente adverso asociado al uso de un dispositivo médico. De otro lado, dentro de las políticas de calidad de la prestación de los servicios de salud, el Ministerio de Salud y Protección Social ha definido la política de seguridad centrada en el paciente, donde el reporte de eventos e incidentes adversos asociados a dispositivos médicos es un importante elemento de articulación de la vigilancia en salud y la prestación de servicios. 2. OBJETIVO Fortalecer la protección de la salud y la seguridad de los pacientes, operadores y todas aquellas personas que se vean implicadas directa o indirectamente en la utilización de dispositivos médicos, mediante la reducción y control del riesgo asociado a estos.
Transcript of MANUAL Código: M-05-001-EQ - Promedan IPS

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
1. JUSTIFICACION Los dispositivos médicos son sometidos a diferentes controles durante su desarrollo, con el fin de garantizar la seguridad y efectividad durante su uso. No obstante, estos controles no son suficientes para garantizar que en la etapa pos-mercado no se presenten problemas o incidentes que puedan desencadenar daños para quienes los utilizan. Por este motivo, existe un consenso mundial sobre la necesidad de sistemas de regulación y vigilancia sobre todo el ciclo de vida de estas tecnologías. Para el caso de Colombia, el país cuenta con una regulación actualizada donde el Ministerio de Salud y Protección Social, el INVIMA, las Entidades Territoriales de Salud, los fabricantes e importadores de dispositivos médicos, tienen la responsabilidad de garantizar que este tipo de tecnologías cumplan con las condiciones clínicas de seguridad, eficacia y desempeño. El INVIMA, en su labor de inspección, vigilancia y control sobre los productos de su competencia, y en cumplimiento de las funciones descritas por la normatividad sanitaria vigente, lidera desde el 2008 el Programa Nacional de Tecnovigilancia. El objetivo principal de este programa es mejorar la protección de la salud y la seguridad de pacientes, usuarios y otros actores involucrados, mediante el control y reducción del riesgo asociado al uso de los dispositivos médicos comercializados en el territorio colombiano. En este sentido, el Instituto ha establecido los mecanismos para recolectar, evaluar y gestionar la información relacionada con la seguridad de los dispositivos médicos, con el propósito de tomar las medidas a que haya lugar para el cumplimiento de este objetivo. Es responsabilidad social y ética de todas las personas involucradas en la fabricación, comercialización, distribución, prescripción, manipulación y uso de los dispositivos médicos informar a la autoridad sanitaria cuando se tenga conocimiento sobre algún evento o incidente adverso asociado al uso de un dispositivo médico.
De otro lado, dentro de las políticas de calidad de la prestación de los servicios de salud, el Ministerio de Salud y Protección Social ha definido la política de seguridad centrada en el paciente, donde el reporte de eventos e incidentes adversos asociados a dispositivos médicos es un importante elemento de articulación de la vigilancia en salud y la prestación de servicios.
2. OBJETIVO Fortalecer la protección de la salud y la seguridad de los pacientes, operadores y todas aquellas personas que se vean implicadas directa o indirectamente en la utilización de dispositivos médicos, mediante la reducción y control del riesgo asociado a estos.

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
3. ALCANCE Este procedimiento aplica a todos los dispositivos empleados en la prestación del servicio de la institución desde la selección, adquisición, almacenamiento, disposición y gestión del uso de los equipos biomédicos en la institución. Aplica para todo el personal que está involucrado en la atención del paciente tanto asistencial como administrativa, en los procesos de recepción, uso y cuidado de pacientes con dispositivos médicos. 4. MARCO LEGAL
Resolución 2003 del 2014, Por la cual se definen los procedimientos y condiciones de inscripción de los Prestadores de Servicios de Salud y de habilitación de servicios de salud.
Decreto 2200 del 2005 por el cual se reglamenta el servicio farmacéutico y se dictan otras disposiciones. Resolución 434 y anexos “Por la cual se dictan normas para la evaluación e importación de tecnologías biomédicas, se define las de importación controlada y se dictan otras disposiciones”. Decreto 4725 del 26 de Diciembre de 2005 por la cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de Dispositivos Médicos para uso humano. Ministerio de la Protección Social. Resolución 4816 de Noviembre 27 de 2008 por la cual se reglamenta el Programa Nacional de Tecnovigilancia. Ministerio de la Protección Social. Resolución 2183 de2004 Definición de Dispositivo Médico, Decreto 4562 de 2006 Vigencias y Control de Mercado a los Dispositivos Médicos. Resolución 2434 de 2006 Por la cual se reglamenta la importación de equipo biomédico repotenciado Clases IIB y III. Resolución 4002 de 2007 Por la cual se adopta el Manual de Requisitos de Capacidad de Almacenamiento y/o Acondicionamiento para Dispositivos Médicos.La ley 9 de 1979 y el Decreto 1562 de 1984, establecen la obligatoriedad para los profesionales y empresas del sector salud, de reportar a las autoridades hechos o eventos, que pongan en riesgo la salud pública. Decreto 4957 de 2007 Por el cual se establece un plazo para la obtención del registro sanitario o permiso de comercialización de algunos dispositivos médicos para uso humano y se dictan otras disposiciones. Decreto 4562 del 2007. Modifica plazo RS para algunos DM. 1 Abril 2007. R-2183 de 2004, Buenas prácticas de esterilización.

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
5. MARCO CONCEPTUAL Acción correctiva: Acción que elimina la causa de un evento adverso u otra situación no deseada, la cual debe ser tomada con el fin de prevenir la recurrencia del evento adverso. Acción Preventiva: Acción que previene la ocurrencia del evento o incidente adverso. Daño: Perjuicio que afecta la salud de las personas, por causar lesión transitoria o permanente, enfermedad o muerte. Defectos de Calidad: Cualquier característica física o química del dispositivo médico que está en contra de las especificaciones definidas por el fabricante y que sirvieron de base para la expedición del registro sanitario o permiso de comercialización por parte del Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA, o que impida que el dispositivo médico cumpla de manera segura y efectiva con el uso previsto durante todo su ciclo de vida. Dispositivo medico: Instrumento, aparato, máquina, software, equipo biomédico, u artículo similar o relacionado, utilizado solo o en combinación, incluyendo sus componentes y programas informáticos, que intervengan en su correcta aplicación, destinado por el fabricante para uso en seres humanos, en los siguientes casos: Diagnóstico, prevención, supervisión, tratamiento o alivio de una enfermedad, investigación, sustitución, modificación o soporte de la estructura anatómica o de un proceso fisiológico, diagnóstico, prevención, supervisión, tratamiento alivio o compensación de una lesión o de una deficiencia (por ejemplo, suturas, ), productos para desinfección y/o esterilización de dispositivos médicos (ejemplo desinfectante) los cuales no ejercen la acción principal que se desea por medios farmacológicos. Dispositivo medico clase I: Son aquellos dispositivos médicos de bajo riesgo, sujetos a controles generales, no destinados para proteger o mantener la vida o para un uso de importancia especial en la prevención del deterioro de la salud humana y que no representan un riesgo potencial no razonable de enfermedad o lesión. Dispositivo medico clase IIA: Son los dispositivos médicos de riesgo moderado, sujetos a controles especiales en la fase de fabricación para demostrar su seguridad y efectividad. Dispositivo medico clase IIB: Son los dispositivos médicos de riesgo alto, sujetos a controles especiales en el diseño y fabricación para demostrar su seguridad y efectividad. Dispositivo medico clase III: Son los dispositivos médicos de muy alto riesgo sujetos a controles especiales, destinados a proteger o mantener la vida o para un uso de importancia sustancial en la prevención del deterioro de la salud humana, o si su uso presenta un riesgo potencial de enfermedad o lesión. Equipo biomédico: Dispositivo medico operacional y funcional que reúne sistemas y subsistemas eléctricos, electrónicos e hidráulicos, incluidos los programas informáticos, que intervengan en su buen funcionamiento, destinados por el fabricante a ser usados en seres humanos con fines de prevención, diagnóstico, tratamiento o rehabilitación.

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Evento adverso por dispositivo medico: Daño no intencionado al paciente, operador o medio ambiente que ocurre como consecuencia de la utilización de un dispositivo médico. Evento adverso por dispositivo medico serio: Evento no intencionado de características irreversibles que pudo haber llevado a la muerte o al deterioro serio de la salud del paciente, una disminución permanente de una función corporal o pérdida permanente de una estructura corporal. Se considera como deterioro serio de la salud: Enfermedad o daño que amenace la vida, daño de una función o estructura corporal, condición que requiera una intervención médica o quirúrgica para prevenir un daño permanente de una estructura o función corporal, evento que lleve a una incapacidad permanente parcial, evento que necesite una hospitalización o una prolongación en la hospitalización, evento que sea el origen de una malformación congénita. Evento adverso por dispositivo médico no serio: Evento no intencionado, diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente, operador o todo aquel que se vea implicado directa o indirectamente, como consecuencia de la utilización de un dispositivo o aparato de uso médico. Pueden ser: Serios: Es aquella condición de característica reversible, que requiere una intervención médica o quirúrgica para prevenir una lesión permanente de una función o la perdida estructural corporal. No serios: Eventos adversos menores que no requieren tratamiento médico y se incluyen los detectados previamente a su uso. Factor de riesgo. Situación, característica o atributo que condiciona una mayor probabilidad de experimentar un daño a la salud de una o varias personas. Incidente adverso por dispositivo medico: Potencial daño no intencionado al paciente, operador o medio ambiente que ocurre como consecuencia de la utilización de un dispositivo médico. Incidente adverso por dispositivo medico serio: Potencial riesgo de daño no intencionado que pudo haber llevado a la muerte o al deterioro serio de la salud del paciente, pero que por causa del azar o la intervención de un profesional de la salud u otra persona, o una barrera de seguridad, no generó un desenlace adverso. Incidente adverso por dispositivo médico no serio: Potencial riesgo de daño no intencionado diferente a los que pudieron haber llevado a la muerte o al deterioro serio de la salud del paciente, pero que por causa del azar o la intervención de un profesional de la salud u otra persona, o una barrera de seguridad, no generó un desenlace adverso. Señal de alerta. Situación generada por un caso o un número de casos reportados con una misma asociación o relación causal entre un evento adverso y un dispositivo médico, siendo desconocida o no documentada previamente y que presuma un riesgo latente en salud.

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Red de Tecnovigilancia. Estrategia nacional de comunicación voluntaria y de trabajo colectivo, que busca articular, apoyar y coordinar el desarrollo de la Tecnovigilancia en Colombia, a través de la participación y comunicación activa entre cada uno de los integrantes del programa y la entidad sanitaria local o nacional. Riesgo. Posibilidad o probabilidad de que pueda producirse un daño, para el paciente y para el personal que lo manipula. Tecnovigilancia: Es el conjunto de actividades que tienen por objeto la identificación y la calificación de eventos adversos serios e indeseados producidos por los dispositivos médicos, así como la identificación de los factores de riesgo asociados a estos efectos o características, con base en la notificación, registro y evolución sistemática de los efectos adversos de los dispositivos médicos, con el fin de determinar la frecuencia, gravedad e incidencia de los mismos para prevenirse la aparión. Trazabilidad. Se refiere a la capacidad de seguir un dispositivo médico a lo largo de la cadena de suministros desde su origen hasta su estado final como objeto de consumo Vigilancia Activa: Se caracteriza por la búsqueda permanente de Eventos e Incidentes adversos relacionados con la utilización de dispositivos médicos Vigilancia Pasiva: Se realiza a través de la notificación de eventos e incidentes adversos relacionados con la utilización de un dispositivo médico. 6. DESARROLLO DEL PROGRAMA Programa Nacional de Tecnovigilancia Está constituido por el conjunto de instituciones, normas, mecanismos, procesos, recursos financieros, técnicos y de talento humano que interactúan para la identificación, recolección, evaluación, gestión y divulgación de los eventos o incidentes adversos no descritos que presentan los dispositivos médicos durante su uso, la cuantificación del riesgo y la realización de medidas en salud pública, con el fin de mejorar la protección de la salud y la seguridad de los pacientes, usuarios y todo aquel que se vea implicado directa o indirectamente con la utilización del dispositivo. Niveles de operación del Programa Nacional de Tecnovigilancia
− A nivel Nacional está integrado por el Ministerio de Salud y Protección Social y el Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA.
− A Nivel Departamental y Distrital, integrado por las diferentes Secretarías Departamentales
− A Nivel Local, integrado por los fabricantes e importadores de dispositivos médicos, Prestadores de Servicios de Salud (IPS), profesionales independientes y los

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
usuarios de dispositivos médicos o cualquier persona que tenga conocimiento de un evento o incidente adverso con dispositivos médicos para uso en humanos.
Representante del programa en la organización Se define como representantes del Programa de Tecnovigilancia, la coordinación Biomédica de la institución en todo lo relacionado con el manejo y funcionamiento de Equipos Biomédicos, y en lo relacionado con la vigilancia y el reporte de eventos adversos asociados a los demás dispositivos médicos, la Química Farmacéutica Asistencial Las funciones de los representantes de este programa son:
− Registrar, analizar y gestionar todo evento o incidente adverso susceptible de ser causado por un dispositivo médico.
− Recomendar medidas preventivas para tomar acciones inmediatamente ocurrido el evento.
− Orientar a los informantes en el correcto diligenciamiento del formato de reporte.
− Sensibilizar a los usuarios de la organización en el Programa de Tecnovigilancia sobre la seguridad y uso adecuado de dispositivos médicos.
− Informar de manera inmediata a la entidad gubernamental que corresponda (Secretarias de salud departamental, Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA, entre otros), todo reporte de evento o incidente adverso serio cuando es del caso de acuerdo a lo establecido en la resolución 4816 de 2008.
− Enviar trimestralmente los informes periódicos a la entidad gubernamental que corresponda de todos los reportes de evento o incidente adverso no serio, de acuerdo a lo establecido en la resolución 4816 de 2008.
− Revisión mensual de alarmas sanitarias, registro, notificación y gestión correspondiente de acuerdo a las recomendaciones del INVIMA.
− Ser el corresponsal del Programa Institucional de Tecnovigilancia ante el gobierno.
− Sensibilizar al conjunto de usuarios y potenciales reportantes de su organización en el Programa de Tecnovigilancia, la seguridad y uso adecuado de dispositivos médicos.
− Enviar trimestralmente los informes periódicos al INVIMA y a la secretaria departamental de todo reporte de evento o incidente adverso no serio, de acuerdo a lo establecido por el análisis de casos.
− Comunicar al fabricante o importador del dispositivo médico correspondiente, la ocurrencia del evento e incidente adverso, si se estima pertinente
− Comunicar al INVIMA y a la secretaria departamental la ocurrencia de eventos e incidentes adversos con el formato de Reporte de riesgo de incidentes adversos a Dispositivos Médicos.
Responsabilidad de la IPS
− Estar atentos y vigilantes del desempeño, calidad y seguridad de los dispositivos médicos previos a su uso.

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
− Informar, divulgar y aplicar las prácticas adecuadas de la utilización de dispositivos médicos.
− Implementar el Programa Institucional de Tecnovigilancia, que asegure un permanente seguimiento de los eventos e incidentes adversos que puedan causar los dispositivos médicos durante su uso y que permitan identificar, registrar, evaluar y gestionar los reportes de eventos e incidentes adversos con los dispositivos médicos que use.
− Tomar las acciones preventivas o correctivas que sean del caso y las que le sean exigidas por el Instituto Nacional de Medicamentos y Alimentos, INVIMA, de forma inmediata.
− Desarrollar actividades de promoción y formación con los profesionales de la salud de la IPS, en relación al desarrollo e implementación del Programa Nacional de Tecnovigilancia y la gestión de eventos o incidentes adversos con dispositivos médicos.
− Cooperar y responder rápidamente a cualquier petición del Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA, sobre la seguridad de los dispositivos médicos.
Gestión de reportes ¿Qué se debe reportar?
Cualquier tipo de evento e incidente relacionados con los dispositivos médicos que ocurra o pueda ocurrir al interior de la IPS y esté relacionado directamente con:
− Dispositivo: Toda aquella falla o interferencia de cualquier dispositivo médico, que pueda interferir con el normal desarrollo del procedimiento, y que pueda afectar finalmente la seguridad e integridad del paciente, debe ser reportado
¿Quién debe hacer el reporte?
− Todo ciudadano o todo profesional de la salud que identifique o tenga conocimiento de que un dispositivo medico es sospechoso de producir o aumento el riesgo de producir un incidente adverso en un paciente.
− Persona natural o jurídica responsable del diseño, fabricación, acondicionamiento y etiquetado de un dispositivo médico para ser comercializado en su propio nombre o de un titular autorizado, independientemente de que algunas de estas operaciones sean realizadas por esta misma persona o por un tercero contratado.
− El titular del registro sanitario, fabricante y/o importador o representante del dispositivo médico, debe notificar al INVIMA todos los problemas, incidentes o riesgo de incidentes adversos relacionado con los dispositivos médicos.
Tipos de reporte
− Reporte inmediato. En caso de presentarse un evento adverso serio con los dispositivos médicos para uso en humanos, Se realiza el reporte de dicho incidente,

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
dentro de las setenta y dos horas (72) horas siguientes a la ocurrencia del evento o incidente.
− Reporte Periódico.Los reportes periódicos se envían trimestralmente y en forma consolidada al INVIMA o a las Dirección Seccional de Salud, de los reportes de eventos e incidentes adversos no serios.
− Reportes bajo el formato de eventos e incidentes adversos: El Personal Asistencial deberá reportar en el formato manual los eventos e incidentes adversos que se presenten agregando a la descripción del evento: Nombre del dispositivo médico, número de registro INVIMA, distribuidor, lote, modelo, serial, placa, marca.
Recomendaciones para el reporte obligatorio:
− Reporte ante la mínima sospecha que el DISPOSITIVO MEDICO pueda ser un factor contribuyente a la ocurrencia de un evento adverso según el cuadro clínico del paciente.
− Reporte toda sospecha de evento- incidente adverso, especialmente los eventos adversos SERIOS donde se sospeche que hay una asociación con el uso del dispositivo médico. Un evento es serio siempre que el Medico lo considere y cuando el paciente: Muere, está o estuvo en riesgo de morir, fue hospitalizado inicialmente o en forma prolongada, presentó una incapacidad (significativa, persistente o permanente), requirió intervención para prevenir lesiones o daños permanentes.
− Reporte los problemas del producto relacionados con: Calidad e integridad de la presentación, sospecha de contaminación, inestabilidad, defectos en sus componentes, efectos en etiquetas e instructivos, calibración y mantenimiento
− Se considera que el reporte se encuentra completo y es útil si contiene la siguiente información: Datos del paciente y dispositivo medico sospechoso, descripción de la(s) sospecha(s) de evento(s) adversos(s) e identificación del reportante.
− Cómo reportar: Diligencie el formulario con la mayor información disponible, utilice páginas adicionales en blanco si es necesario ampliar información, utilice por cada paciente – dispositivo un formulario, remita el formulario al Responsable del Programa de Tecnovigilancia, reporte aun cuando usted no esté seguro de que el producto causó el evento, no deje de enviar el formulario por carecer de alguna información
− La información contenida en este reporte es información epidemiológica, por lo tanto, tiene carácter confidencial y se utilizará únicamente con fines sanitarios. El Ministerio de la Protección Social y el INVIMA son las únicas instituciones competentes para su divulgación. (Ley 9 de 1979)
7. PROCEDIMIENTO REPORTE Y GESTION DE EVENTOS SUJETOS A TECNOVIGILANCIA
A través de nuestra página web los colaboradores tienen la posibilidad de acceder al software de gestión de eventos asociados a la atención en salud, por medio del cual se pueden notificar los eventos de tipo: adverso, incidente y servicio no conforme, en el aplicativo se deberán registrar los siguientes datos:
− Evento relacionado con la atención clínica del paciente

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
− Daño producido en el paciente
− Listado de principales eventos de seguridad
− Datos básicos del evento: Fecha de ocurrencia, sede, servicio
− Descripción del evento
− Acción inmediata realizada
Ruta: http://www.promedanips.com/ colaboradores/ portal de aplicaciones/ historia clínica Evolution/ usuario/ contraseña/ sede/ seguridad del paciente/ notificación. Formato para notificación de eventos asociados a la atención en salud Después de realizar el debido reporte en el aplicativo de seguridad del paciente, si el evento e incidente tiene relación con un dispositivo medico se debe diligenciar el formato FOREIA001. Este se encuentra disponible para todos los colaboradores y se puede acceder a él a través de la intranet corporativa. Los formatos diligenciados son recolectados semanalmente por los coordinadores o directores de cada servicio o IPS, para luego ser entregados al proceso de seguridad del paciente. En caso de ser un evento grave o centinela, se debe informar inmediatamente al coordinador del servicio y al Gestor de Seguridad del Paciente para su respectiva intervención inmediata. Promedan IPS centra sus esfuerzos en la creación de una cultura de seguridad del paciente, incentivando el reporte voluntario de eventos asociados a la atención en salud con un carácter formativo y no punitivo. NOTA: Para ampliar información, remítase al MANUAL M-02-003-GSP-Monitorizacion aspectos relacionados con SP disponible en la intranet
Tecnovigilancia pasiva
ACTIVIDAD DESCRIPCION RESPONSABLE
Detección del incidente o evento adverso
Identificar el evento adverso o incidente por Dispositivo Medico.
Personal Asistencial
Tomar medidas preventivas o correctivas pertinentes
Tomar las medidas pertinentes para mejorar la situación clínica del paciente en caso que la salud de éste haya sido afectada por un evento adverso por dispositivo médico.
Personal Asistencial
Registro de la información para realizar el reporte.
En el reporte de eventos e incidentes adversos, el Personal Asistencial deberá reportar en el aplicativo de seguridad del paciente, el caso identificado agregando a la descripción los siguientes datos: Nombre del dispositivo, número de registro INVIMA, distribuidor, lote, modelo, serie, marca y placa.
Personal Asistencial Representante Asistencial del programa

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
ACTIVIDAD DESCRIPCION RESPONSABLE
El representante del programa ante la red nacional deberá diligenciar el “Formato de reporte de evento incidente adverso con dispositivo médico-FOREIA001” definido por el INVIMA. Al diligenciar cada uno de los campos del formulario incluya datos completos, dado que la carencia de datos como el número de Registro Sanitario, lote, modelo, referencia o serial del dispositivo médico, no permitirá iniciar la investigación. NOTAS: Si en el evento o incidente adverso se encuentra involucrado más de un (1) dispositivo médico sospechoso, diligencie un formulario por cada uno de ellos, identificándolos como dispositivo 1, dispositivo 2 etc. Utilice hojas adicionales cuando los espacios establecidos en el formulario no resulten suficientes para describir en forma clara y concisa el evento o incidente adverso, corrobore que incluya información respecto a cómo se detectó y las medidas tomadas. De ser posible, adjunte certificados, dibujos, fotografías o copias de folletos, catálogos, instrucciones de uso o manuales; con frecuencia esta documentación es de suma utilidad para describir e interpretar, las circunstancias en las que se produjo el evento o incidente. Las fechas deberán ser ingresadas como dd/mm/aaaa (ej. Febrero 3 de 2012 = 03/02/2012). Si desconoce la fecha exacta, diligencie la más aproximada.
Envío del Reporte Primario
Envíe el reporte diligenciado al responsable del Programa de Tecnovigilancia.
Personal Asistencial
Recepción del reporte
Recepción del reporte diligenciado y verifique que el mismo contenga la información necesaria para la evaluación del evento adverso por dispositivo médico: número de Registro Sanitario, lote, modelo, referencia o serial del dispositivo médico,
Representante Asistencial del Programa

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
ACTIVIDAD DESCRIPCION RESPONSABLE
Revisión del caso
Hacer una revisión bibliográfica o una entrevista con el personal que identifica evento/ incidente
Representante Asistencial del Programa
Análisis de los reportes
Identifique los eventos adversos y/o incidentes adversos por dispositivos médicos, analice sus posibles causas y sugiera acciones preventivas y correctivas, con base en los reportes entregados. Para el análisis e investigación se utilizará como metodología “Protocolo de Londres”. Si el caso está relacionado con equipos biomédicos, solicite el apoyo del técnico idóneo encargado del mantenimiento de dichos equipos para el análisis. Se propondrán acciones preventivas o correctivas si se determina que el evento adverso fue evitable.
Representante Asistencial del Programa

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
ACTIVIDAD DESCRIPCION RESPONSABLE
Envió de los reportes al INVIMA y a la DSSA
Enviar el formulario FOREIA001 al INVIMA en caso de presentarse un evento adverso SERIO, dentro de las setenta y dos horas (72) siguientes al conocimiento de la ocurrencia del mismo. Este se ingresa en la página Web del INVIMA en la opción de reportes FOREIA, puede ser enviado al correo electrónico [email protected], o por correo físico a la carrera 10 No. 64-28 Bogotá. Si el caso corresponde a un evento adverso NO SERIO, se consolida la información en el formato RETEIM-002, y se realiza el cargue de la información contenida en la página web https://farmacoweb.invima.gov.co/TecnoVigilancia/, se ingresa con el usuario y la contraseña de la institución.
Recuerde comunicar al fabricante o importador del dispositivo médico correspondiente, la ocurrencia del evento e incidente adverso e informar el código de identificación interno
Representante Asistencial del Programa

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
ACTIVIDAD DESCRIPCION RESPONSABLE
asignado al reporte por parte del INVIMA, a fin de mejorar la identificación, recolección y la gestión de cada caso. Si durante el trimestre no se presentaron eventos e incidentes adversos asociados al uso de dispositivos medicos, se procederá a realizar el Reporte Trimestral en Cero en el aplicativo Web del INVIMA.
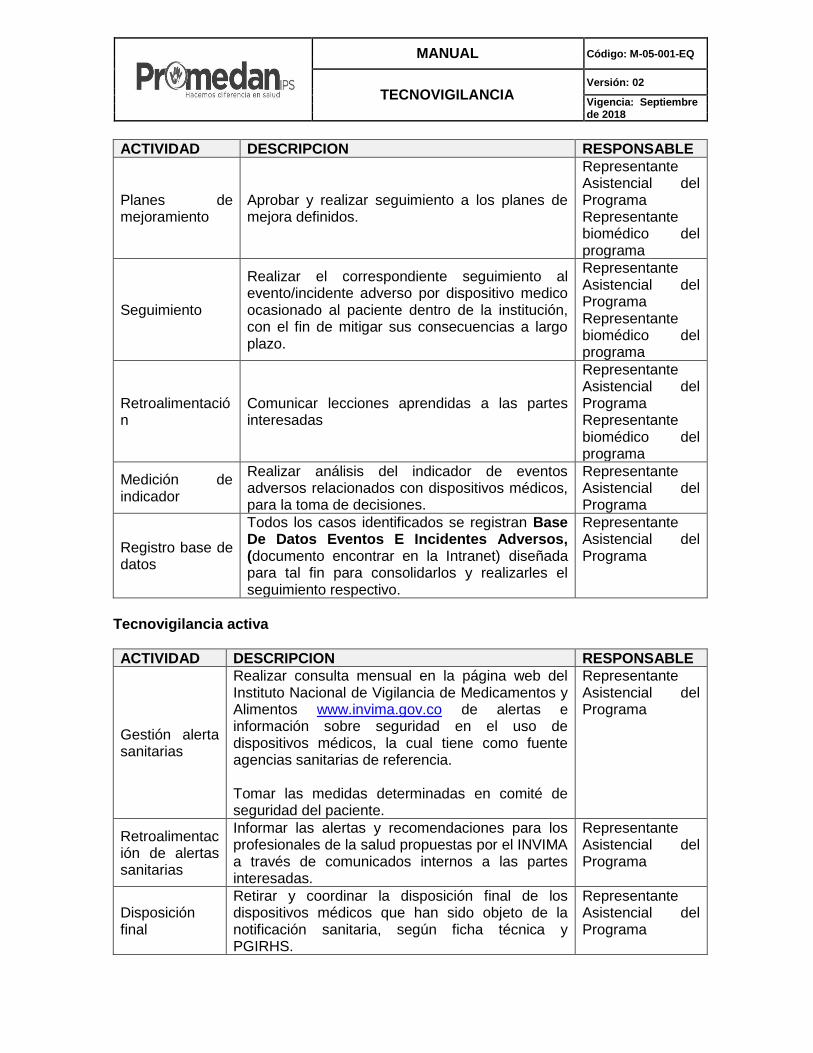
MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
ACTIVIDAD DESCRIPCION RESPONSABLE
Planes de mejoramiento
Aprobar y realizar seguimiento a los planes de mejora definidos.
Representante Asistencial del Programa Representante biomédico del programa
Seguimiento
Realizar el correspondiente seguimiento al evento/incidente adverso por dispositivo medico ocasionado al paciente dentro de la institución, con el fin de mitigar sus consecuencias a largo plazo.
Representante Asistencial del Programa Representante biomédico del programa
Retroalimentación
Comunicar lecciones aprendidas a las partes interesadas
Representante Asistencial del Programa Representante biomédico del programa
Medición de indicador
Realizar análisis del indicador de eventos adversos relacionados con dispositivos médicos, para la toma de decisiones.
Representante Asistencial del Programa
Registro base de datos
Todos los casos identificados se registran Base De Datos Eventos E Incidentes Adversos, (documento encontrar en la Intranet) diseñada para tal fin para consolidarlos y realizarles el seguimiento respectivo.
Representante Asistencial del Programa
Tecnovigilancia activa
ACTIVIDAD DESCRIPCION RESPONSABLE
Gestión alerta sanitarias
Realizar consulta mensual en la página web del Instituto Nacional de Vigilancia de Medicamentos y Alimentos www.invima.gov.co de alertas e información sobre seguridad en el uso de dispositivos médicos, la cual tiene como fuente agencias sanitarias de referencia. Tomar las medidas determinadas en comité de seguridad del paciente.
Representante Asistencial del Programa
Retroalimentación de alertas sanitarias
Informar las alertas y recomendaciones para los profesionales de la salud propuestas por el INVIMA a través de comunicados internos a las partes interesadas.
Representante Asistencial del Programa
Disposición final
Retirar y coordinar la disposición final de los dispositivos médicos que han sido objeto de la notificación sanitaria, según ficha técnica y PGIRHS.
Representante Asistencial del Programa

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Seguimiento factores ambientales
Controlar las condiciones de las áreas de almacenamiento (temperatura y humedad relativa) recomendadas por el fabricante.
Personal Asistencial
Control fechas de vencimiento
Verificar continuamente la fecha de vencimiento de los dispositivos médicos (cuando aplique) y validar con calidad la destrucción. De acuerdo con lo establecido en la ficha técnica y el PGIRHS.
Personal Asistencial
Seguimiento a Calibraciones
Verificar el estado de vigencia de la calibración de dispositivos médicos cuando aplique, según Programa de Aseguramiento Metrológico.
Representante biomédico del programa
Gestión de capacitaciones
Realizar entrenamiento y reentrenamiento de los usuarios con las casas comerciales para garantizar el uso correcto, seguro y eficaz de los equipos y su software.
Representante Asistencial del Programa Representante biomédico del programa
Seguimiento a instalaciones
Verificar que el formato de mantenimiento del equipo entregado por la casa comercial cumpla con los siguientes requisitos: -Nombre del Equipo -Ubicación del Equipo -Tipo de Mantenimiento realizado (Cumplimiento del protocolo de mantenimiento) -Estado de Entrega del Equipo: "Equipo en condiciones óptimas para su uso" -Fecha en que se realizó. De acuerdo al cronograma de mantenimientos preventivos. -Nombre de quién recibe a conformidad
Representante biomédico del programa
Seguimiento mantenimientos preventivos
Realizar seguimiento al cumplimiento del cronograma de mantenimientos preventivos de los equipos biomédicos.
Representante biomédico del programa
Priorización de dispositivos médicos
Establecer el grupo de dispositivos médicos objeto de vigilancia activa en comité de seguridad del paciente, teniendo en cuenta el historial de incidentes y/o eventos adversos u otros temas definidos por la Institución
Representante Asistencial del Programa
Educación al usuario
Suministrar información al paciente sobre el uso y riesgos del uso de los dispositivos médicos; haciendo participe al paciente de su propia seguridad. La adherencia es verificada en las rondas de seguridad. En caso de presentarse un incidente o evento adverso con alguno de los dispositivos médicos priorizados proceder a realizar reporte teniendo en cuenta el procedimiento de tecnovigilancia pasiva
Representante Asistencial del Programa

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Análisis y Seguimiento
Analice sus posibles causas y sugiera acciones preventivas y correctivas, con base en los reportes entregados. Para el análisis e investigación se utilizará como metodología “Protocolo de Londres”. Si el caso está relacionado con equipos biomédicos, solicite el apoyo del técnico idóneo encargado del mantenimiento de dichos equipos para el análisis. Determinar si las barreras de seguridad establecidas no fueron suficientes: Se deben reforzar o crear nuevas.
Representante Asistencial del Programa
Retroalimentación al grupo asistencial
Informar al grupo de salud los resultados del análisis y seguimiento. Instruir sobre las nuevas barreras de seguridad establecidas.
Representante Asistencial del Programa
8. GESTION DE ALERTAS EMITIDAS POR EL INVIMA: Un responsable asignado por el comité de Tecnovigilancia mensualmente realiza una búsqueda sistemática de las alertas emitidas por Organismos Reguladores Internacionales mediante monitoreo de la página oficial del INVIMA, a través de la siguiente ruta: www.invima.gov.co/Tecnovigilancia/Gestión de Informes de Seguridad - Alertas - Recalls e Hurtos (Risarh)/Alertas Internacionales. La información que se puede encontrar en las páginas oficiales contempla recomendaciones, información general, Alertas sobre productos como también los retiros de producto del mercado notificado por los fabricantes de dispositivos médicos en cumplimiento de sus responsabilidades respecto a la seguridad de los productos que comercializa. El registro de la consulta de estas alertas y la gestión que se le realiza se encuentra plasmada en el formato FT-05-002-EQ- Seguimiento Alertas Sanitarias, como también el reporte de los Hurtos de los DM, formato FT-05-003-EQ- Hurtos Dispositivos Médicos. De igual manera se puede El seguimiento a esta actividad se realiza mes vencido en el comité de riesgo clínico.
9. SEGUIMIENTO A DISPOSITIVOS MEDICOS IMPLANTABLES
El seguimiento a dispositivos médicos implantables se realiza a través de un sistema implementado llamado “Triple Tarjeta”, el cual permite realizar la trazabilidad de los dispositivos médicos implantados a partir del registro de la información del dispositivo médico en un formato. Es conocido como “triple tarjeta” debido a la distribución de las fichas: una para el paciente, una para la historia clínica del paciente y otra para el proveedor del dispositivo.

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
(Artículo 63°-Decreto 4725 del 2005, Articulo 31-Resolución 4816 del 2008) El registro de la información del dispositivo médico implantado se realiza en la historia clínica por el personal de enfermería. La información registrada en la triple tarjeta corresponde a: - Nombre completo del paciente - Identificación - Dirección - Teléfono - Sexo - Procedimientos - Médico que realiza el procedimiento - Institución - Fecha del procedimiento - Diagnostico - Nombre del DM - Lote o serie - Registro sanitario - Fabricante - Dirección del fabricante - Cantidades
El reporte de la “triple tarjeta” se reporta a los proveedores de dichos dispositivos trimestralmente por el líder del comité de tecnovigilancia (Agosto, Noviembre, Febrero, Mayo), a nivel hospitalario las enfermeras diligencian en SERVINTE la triple tarjeta, con el fin de implementar la base de datos de los dispositivos médicos implantados en la Clínica, desde los servicios farmacéuticos satélites se registra la información de la triple tarjeta de la historia clínica en el formato Trazabilidad de dispositivos médicos implantados (anexo 5), encontrado en la intranet, y este es enviado al encargado del servicio farmacéutico diariamente, quien mandara al comité de tecnovigilancia semanalmente dejando plasmada la información en el formato de Trazabilidad de dispositivos medicos implantados, , y se encarga de enviar al proveedor dicha información trimestralmente. En la parte ambulatoria el personal de enfermería dejan plasmada la información directamente en el formato Trazabilidad de dispositivos medicos implantados, y se encarga de mandarlo al correo de tecnovigilancia: [email protected], y el líder de tecnovigilancia se encarga de igual manera de hacerlo llegar al proveedor según el tiempo establecido. La información del dispositivo médico implantado se entrega a cada paciente dependiendo del tipo de atención: los pacientes ambulatorios reciben la “triple tarjeta” desde el quirófano al momento del alta. Los pacientes hospitalizados reciben la información impresa por las secretarias de cada servicio al momento del alta Los dispositivos médicos a los cuales se les realiza trazabilidad son los siguientes:
− Mallas no absorbibles*
− Stent en general
− Material de Osteosíntesis

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
− Catéter JJ
− Válvulas mecánicas y/o biológicas
− Injertos rectos y bifurcados
− Catéter de derivación ventrículo-peritoneal
− Marcapasos
− Neuroestimulador
− Catéter implantable de QT Se consideran Mallas no absorbibles las siguientes:
− Malla gynemesh
− Malla incont. Swing-band ref sb3 10400 dg cardiomed
− Malla incontinencia just-swing svs svs0103010 cardiomed
− Malla mini sling ophira rp
− Malla miniarc
− Malla monarc para incontinencia
− Malla parietene en general
− Malla parietex en general
− Malla polipropileno
− Malla sis direct soft dynamesh
− Malla sistema de suspensión splentis
− Malla sparc
− Malla tvt abbrevo j&j
− Malla ref/nazca tc (para cistocele)
− Malla solyx tecnologias médicas
− Malla surelift para prolapso invivo
10. MEDIDAS PREVENTIVAS PARA EVITAR CASOS DE SEGURIDAD CON DISPOSITIVOS MEDICOS
− Garantizar durante la adquisición que el equipo o el dispositivo médico cumpla con todas las especificaciones técnicas requeridas y la regulación vigente
− Verificar que no existe en el país ninguna alerta relacionada con la utilización de los mismos
− Verificar los registros INVIMA de los equipos y/o dispositivos médicos.
− Para equipos biomédicos, ingresar el equipo en el plan de mantenimiento preventivo definido en la empresa
− Garantizar que los equipos que lo requieran estén incluidos en el programa de calibración y metrología
− Garantizar la disponibilidad de las guías rápidas para equipos biomédicos y que sean de fácil consulta por el personal que opera los equipos.
− Mantener los dispositivos médicos en condiciones adecuadas de almacenamiento y control de fechas de vencimiento.
− Garantizar el entrenamiento de todo el personal de la institución que utilizará el equipo y/o dispositivo médico

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
− Tener disponibles los manuales de entrenamiento y demás especificaciones de uso descritas por el fabricante.
11. BIBLIOGRAFIA
− Instituto Nacional de Vigilancia y Alimentos, INVIMA. www.invima.gov.co
− Decreto 4725 del 26 de Diciembre de 2005 por la cual se reglamenta el régimen de registros sanitarios, permiso de comercialización y vigilancia sanitaria de Dispositivos Médicos para uso humano. Ministerio de la Protección Social.
− Resolución 4816 de Noviembre 27 de 2008 por la cual se reglamenta el Programa Nacional de Tecnovigilancia. Ministerio de la Protección Social.
− ABC de Tecnovigilancia
− ABC de Dispositivos Médicos
13.12. ANEXOS
− Reporte obligatorio de Evento o Incidente Adverso asociado al uso de un Dispositivo Médico FOREIA001
− Reporte Trimestral de eventos adversos no serios con Dispositivos Médicos RETEIM-00
14.13. CONTROL DE CAMBIOS
15.14. ANEXOS
Anexo 1:
Versión Fecha Descripción Elaboró Revisó y Aprobó
1 Septiembre de 2016
Primera versión Gestión de Biomedicina
Gestión de Calidad
2 Septiembre de 2018
Actualización Gestión de Biomedicina
Gestión de Calidad

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Reporte obligatorio FOREIA001

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Anexo 2: Reporte Trimestral RETEIM-002
Anexo 3: Formtato de Seguimiento alertas nacionales e internacionales tecnovigilancia.

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Anexo 4: Formato de notificacion de eventos asociados a la atencion en salud

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Anexo 5: Trazabilidad de DM implantables

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Anexo 6. Proceso para el diligenciamiento de la “triple tarjeta” por parte de enfermería a nivel hospitalario
1. Ingresar al tablero clínico de pacientes (hospitalización o consulta externa según corresponda) 2. Buscar el paciente por nombre o servicio 3. Ingresar a la aplicación de consentimiento informado
4. En la aplicación de consentimiento informado dar clic sobre el botón NUEVO y seleccionar la opción Doc. sin Actividad = SI

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
5. Anotar en el campo documento el código DIPS o seleccionar de la lista desplegable del mismo campo el documento denominado TARJETA DE IMPLANTE DE DISPOSITIVOS:
6. Luego proceder a diligenciar el formato dando doble clic sobre el campo INFORMACIÓN DEL DISPOSITIVO
7. luego de editar la información solicitada en el texto, seleccionar el botón de ACEPTAR, posteriormente GUARDAR y FIRMAR cuando lo solicite.
Anexo 7. Proceso para la consulta de la triple tarjeta por el servicio farmacéutico
1. Ingresar al tablero clínico de pacientes (hospitalización o consulta externa según corresponda)
2. Buscar el paciente por nombre o servicio 3. Ingresar a la aplicación de consentimiento informado

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
4. Ingresar a la aplicación HISTÓRICO 5. Seleccionar la opción ATENCIÓN ENFERMERÍA QUIRÓFANO correspondiente al
día del procedimiento quirúrgico
6. Seleccionar la opción INFORMACIÓN DEL DISPOSITIVO
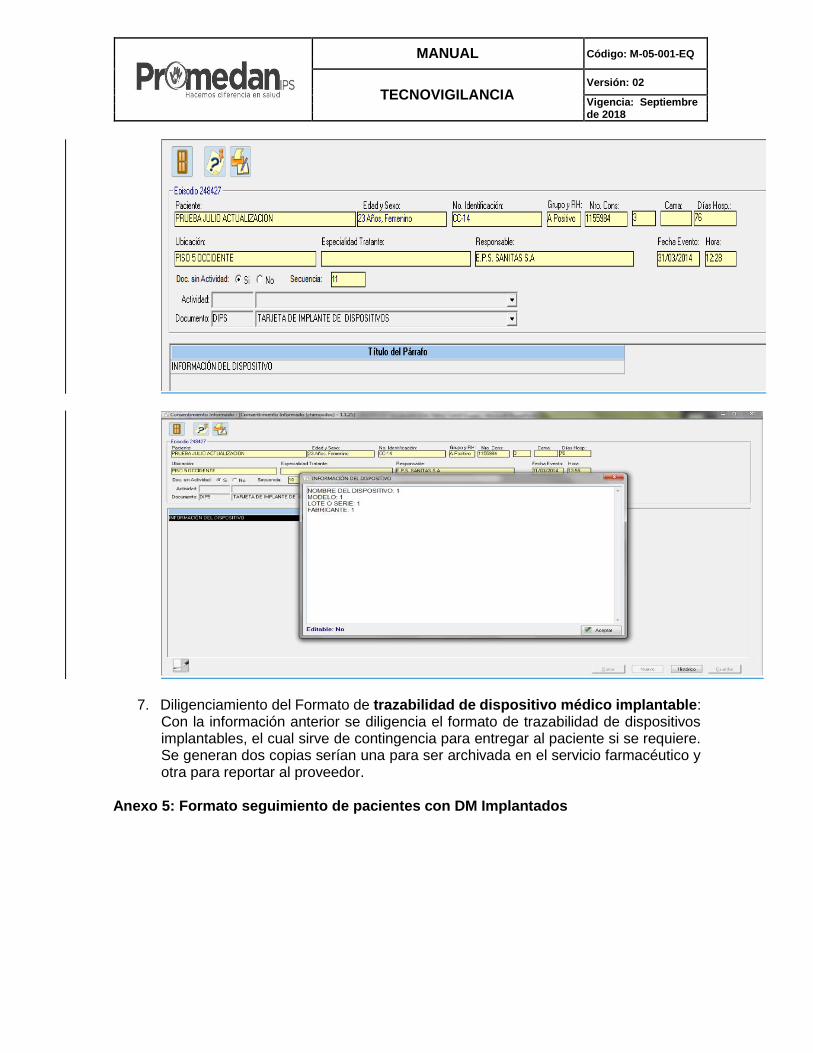
MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
7. Diligenciamiento del Formato de trazabilidad de dispositivo médico implantable: Con la información anterior se diligencia el formato de trazabilidad de dispositivos implantables, el cual sirve de contingencia para entregar al paciente si se requiere. Se generan dos copias serían una para ser archivada en el servicio farmacéutico y otra para reportar al proveedor.
Anexo 5: Formato seguimiento de pacientes con DM Implantados

MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018
Anexo 6: Reporte de eventos e incidentes adversos
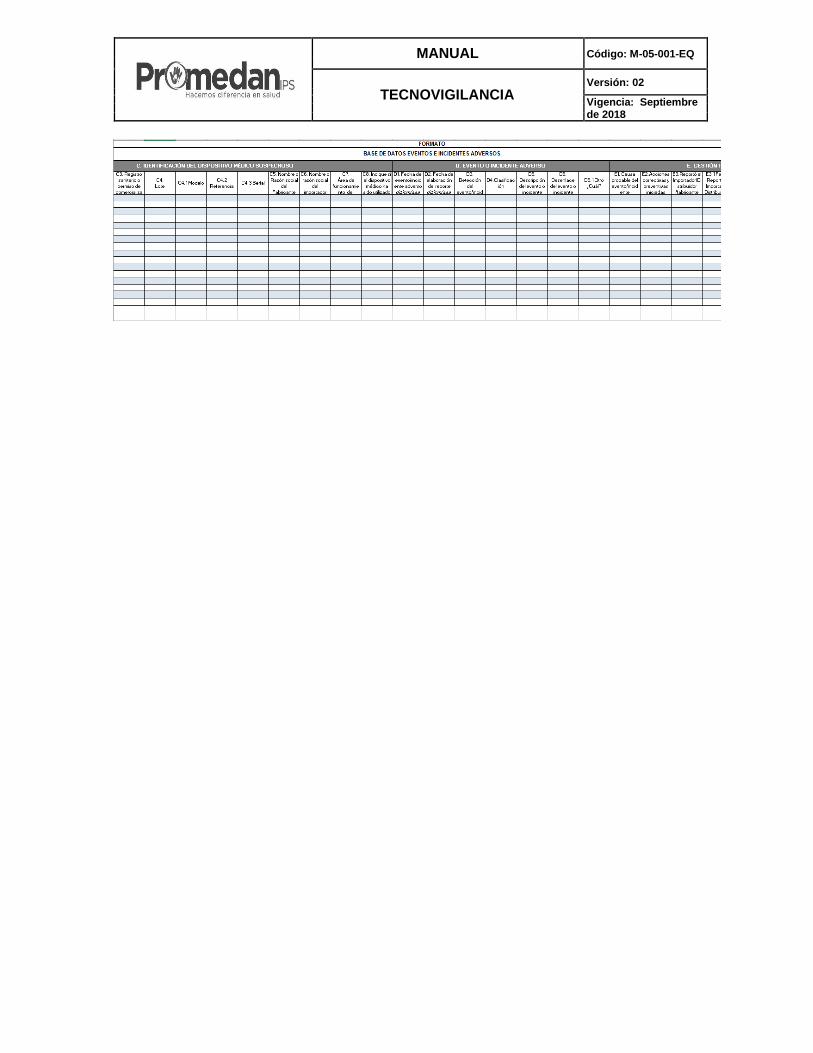
Anexo 7: Base de datos eventos e incidentes adversos
MANUAL Código: M-05-001-EQ
TECNOVIGILANCIA
Versión: 02
Vigencia: Septiembre de 2018


















