Sindromes
99
Enfermedades Integrantes: Barajas Navarro Raúl II. Cuevas Sandoval Virginia Daniela. Lupercio Segoviano Guille María Fernanda. Neri Carrillo Alejandra. Ortega González Jessica Karina
-
Upload
alejandra-neri -
Category
Health & Medicine
-
view
212 -
download
0
Transcript of Sindromes

Enfermedades Integrantes:
Barajas Navarro Raúl II.
Cuevas Sandoval Virginia Daniela.
Lupercio Segoviano Guille María Fernanda.
Neri Carrillo Alejandra.
Ortega González Jessica Karina

Síndrome de Prader-Willi

Antecedentes: Síndrome de Prade-Willi
• Es una enfermedad congénita (presente desde el nacimiento) que afecta muchas partes del cuerpo.

Etiología e incidencia
• Trastorno congénito no hereditario y poco común.
• Mutación puede ser activa e inactiva de forma diferente según de donde procedan ya sea madre o padre por u11 --> imprinting.
• No está relacionando con sexo, raza o condición de vida y su incidencia es aproximadamente de 1 por cada 10,000 nacidos
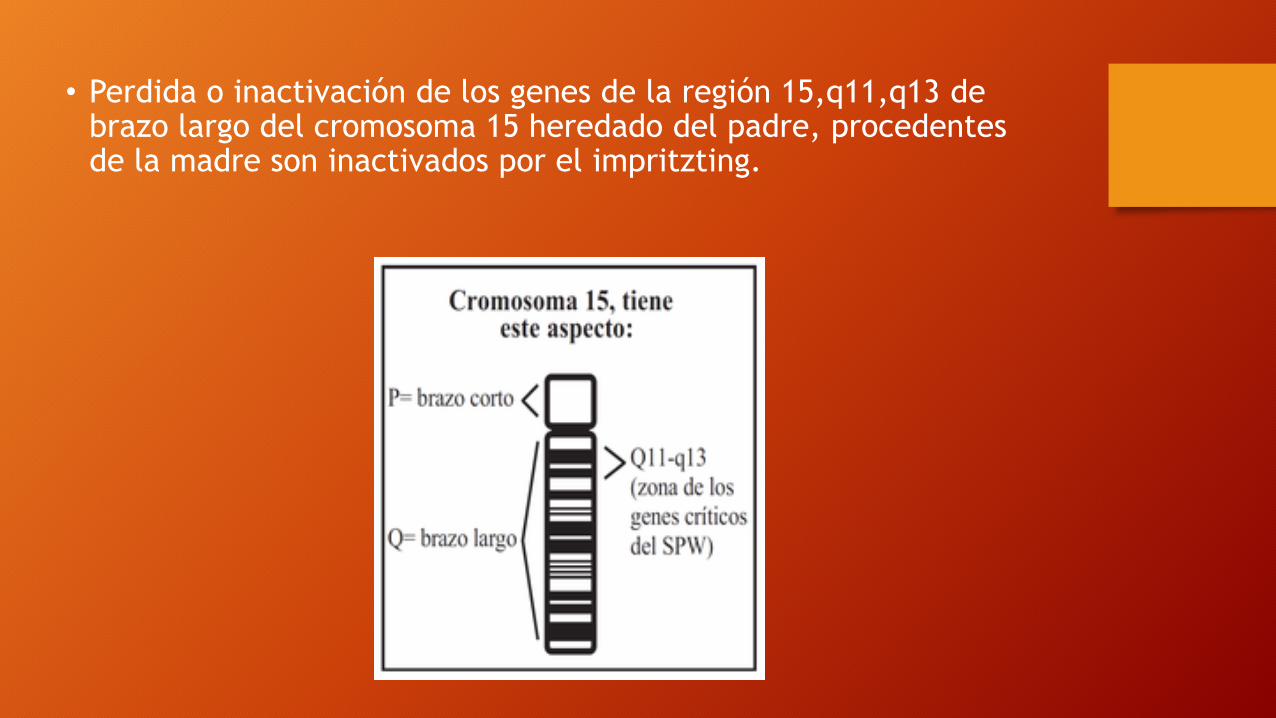
• Perdida o inactivación de los genes de la región 15,q11,q13 de brazo largo del cromosoma 15 heredado del padre, procedentes de la madre son inactivados por el impritzting.

Características clínicas importantes


Trastorno de múltiples etapas: (fases)
Hipotónica: dif. Casos de hipotonía en periodo neonata, y la primera infancia
Llanto débil
Hipotermia
Hipogenitalismo
Pobre reflejo de succión
Durante el primer año, los niños con SPW se definen como amables, tranquilos, cariñosos.
Hiperfagicos: comienza entre 1 y 2 años de vida
Apetito voraz
Hipofagia
Inició precoz de obesidad infantil
Inactividad física
Disminución de la sensibilidad al dolor
Retirado psicomotor
Dificultad al hablar
Disfunción cognitiva
Automutilacion
Impulsividad
Adolescencia y edad adulta: problemas de salud secundarios a obesidad
Escoliosis
Problemas dentales
DM
Hipertensión arterial
Problemas psiquiátricos
Disfunción del hipotálamo
Niveles disminuidos de hormonas sexuales
Órganos sexuales de tamaño insuficiente
Descontrol en la temperatura corporal
Exceso de grasa corporal y déficit de masa magra


Asesorías
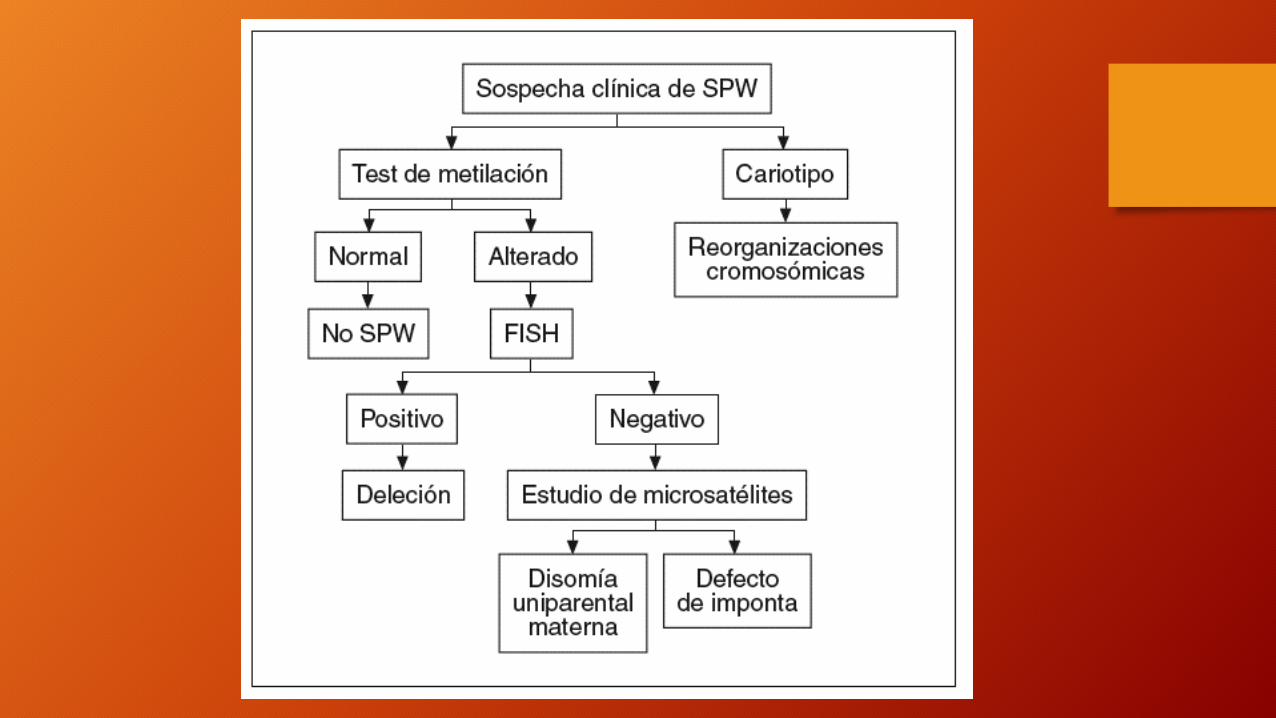
• Consulte con el médico si su hijo presenta síntomas de esta afección. Este trastorno se sospecha frecuentemente al momento del nacimiento.
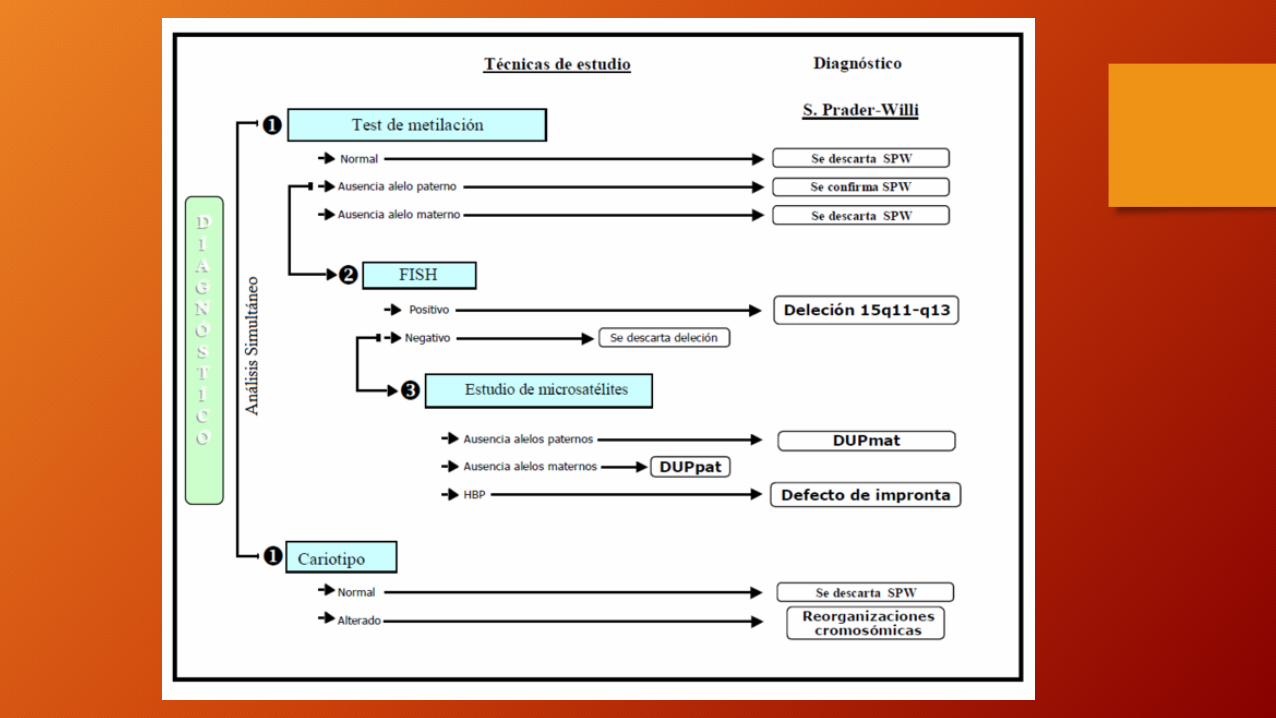

Tratamiento
• La obesidad es el mayor problema --> reducción de las calorías controlará la obesidad.
• ADH:
• Mejora la fortaleza física y la agilidad
• Mejora la estatura
• Incrementa la masa muscular magra y disminuir la grasa corporal
• Mejora la distribución del peso
• Incrementa el vigor
• Incrementa la densidad mineral osea

Tratamiento fisioterapeutico debe iniciarse desde el nacimiento. Ya que se presentan:
• Hipotonia
• Escoliosis
• Marcha
• Fortalecimiento muscular
• Estimulación temprana – terapia ocupacional

Pronostico
• El niño necesitará la educación adecuada para su nivel de CI. También necesitará terapia de lenguaje, terapia ocupacional y fisioterapia lo más pronto posible. El control de peso le permitirá una vida mucho más confortable y saludable.

Síndrome de Angelman

¿Que es el síndrome de Angelman?
• Es una enfermedad genética que causa problemas con la forma como se desarrollan el cerebro y el cuerpo de un niño. El síndrome está presente desde el nacimiento (congénito). Sin embargo, a menudo no se diagnostica hasta los 6 a 12 meses de edad.

Mutación e incidencia
• Estas mutaciones están en una zona del cromosoma 15, en el mismo locus que el síndrome de Prader-Willi, precisamente en el 15q11-q13.
• Tiene una incidencia estimada de un caso cada 15 000 a 30 000 nacimientos
• Ejemplo clásico de enfermedad genética cuyo origen y herencia dependen del mecanismo de impronta genética.

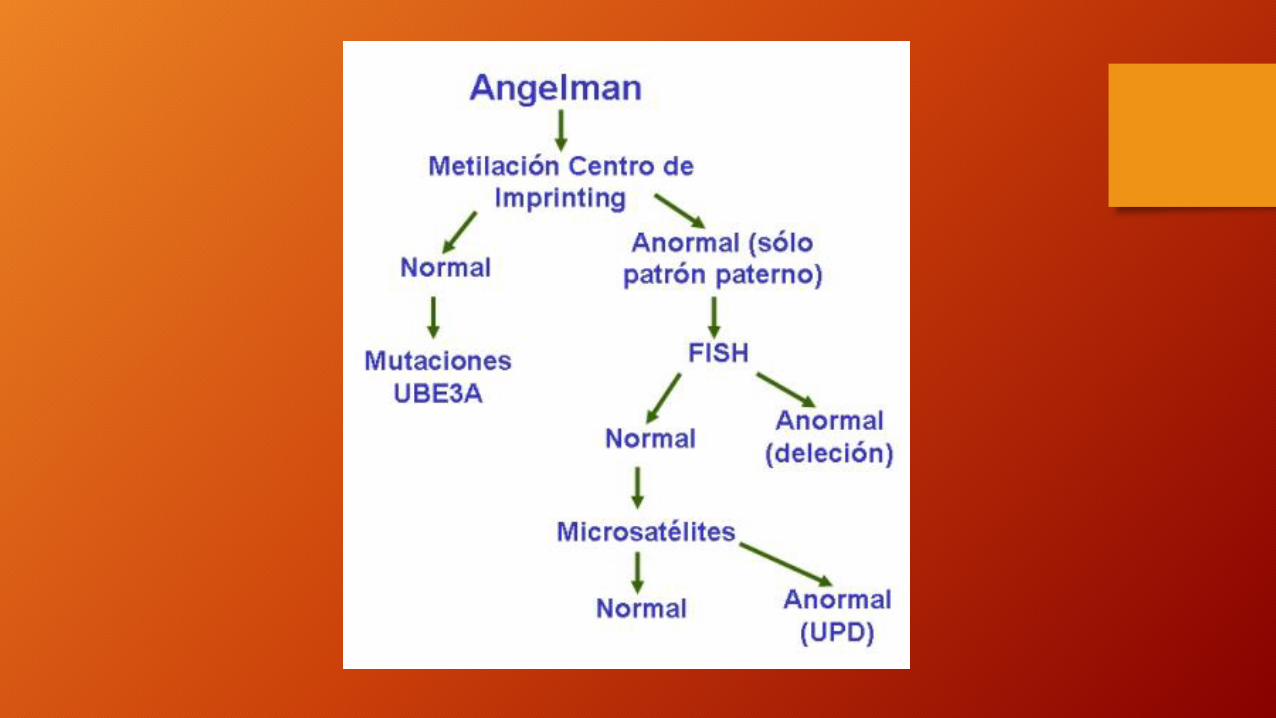
Patrón de herencia
• En el 70% de los niños con síndrome de Angelman, solo se ve el patrón de metilación del cromosoma del padre, indicando esto que la región 15q 11-13 está, o bien totalmente perdida debido a la gran delación materna, o tiene los dos cromosomas 15 de origen paterno, o tiene un defecto en el centro del "Imprinting" .

Características clínicas
En los recién nacidos y lactantes:
Pérdida del tono muscular (flacidez)
Problemas de alimentación
Acidez gástrica (reflujo por acidez)
En niños pequeños y niños mayores:
Marcha espasmódica o inestable
Habla muy poco o no habla para nada
Personalidad feliz y excitable
Reír y sonreír con frecuencia
Cabello, piel y color de los ojos claro en comparación con el resto de la familia
Cabeza de tamaño pequeño en comparación con el cuerpo, parte posterior de la cabeza aplanada
Discapacidad intelectual grave
Convulsiones
Movimiento excesivo de las manos y extremidades
Problemas para dormir
Interposición lingual, babeo
Masticación y movimientos inusuales con la boca
Ojos bizcos
Caminar con los brazos levantados y las manos saludando

Manejo
• No hay ninguna manera de prevenir el síndrome de Angelman. Si usted tiene un niño con este síndrome o un antecedente familiar de la enfermedad, tal vez quiera hablar con el médico antes de quedar embarazada.

Manejo
• No existe cura para el síndrome de Angelman. El tratamiento ayuda a manejar los problemas de salud y del desarrollo causados por la enfermedad.
Anticonvulsivos Terapia conductual Terapia ocupacional y
logopedia
Fisioterapia
Controlar convulsiones Manejar hiperactividad
Problemas de suelo y desarrollo
Problemas del habla
Enseñan habilidades para la
vida
Con problemas de
movimiento y marcha
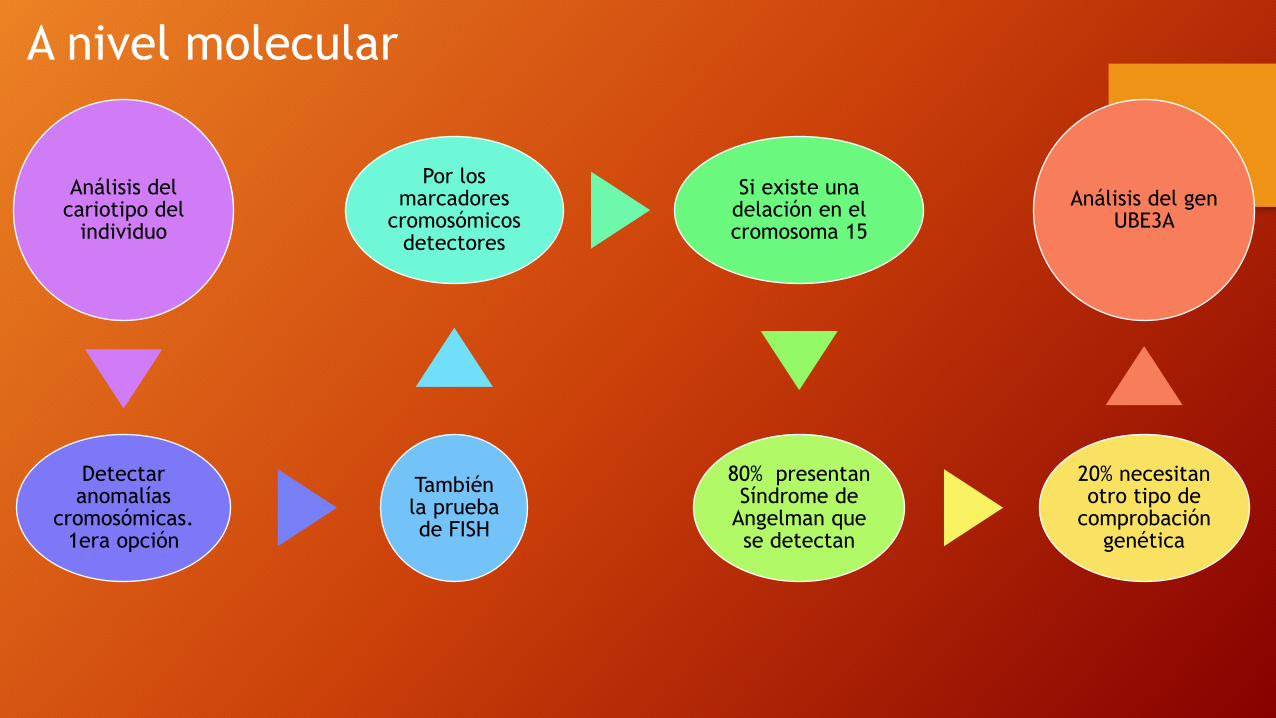
Análisis del cariotipo del
individuo
Detectar anomalías
cromosómicas. 1era opción
También la prueba de FISH
Por los marcadores
cromosómicos detectores
Si existe una delación en el cromosoma 15
80% presentan Síndrome de
Angelman que se detectan
20% necesitan otro tipo de
comprobación genética
Análisis del gen UBE3A
A nivel molecular

Retraso mental Sitio X frágil

Retraso mental Sitio X frágil
• Trastorno hereditario que ocasiona deficiencia mental, pudiendo ser éste de moderado a grave, y siendo la segunda causa genética del mismo, solo superado por el síndrome de down
Etiología
• La herencia es una mutación de tipo dominante ligada a X, aunque no responde a las reglas usuales de dicha herencia, ya que hay varones y mujeres portadoras normales entre la progenie de estas últimas

Signos y síntomas
• Las características principales de este síndrome, han de tenerse muy en cuenta en personas con autismo, deficiencia mental o problemas para el aprendizaje

Fenotipo
• Los rasgos son cara alargada, frente prominente, mentón pronunciado y grandes orejas
• Además presentan articulaciones hiperextensibles, habla reiterativa, testículos grandes, orejas prominentes y bajo tono muscular

Manejo
• No existe un tratamiento
• Se busca un desarrollo educativo y interacción que ayude a los niños afectados

ENFERMEDAD DE HUNTINGTON
• La palabra corea proviene del griego (choreia) que significa danza.
• Este término fue utilizado por primera vez por el médico alquimista Paracelso (1493- 1541) para describir la Corea o ¨baile de San Vito¨.
• La EH, conocida también como el “mal de San Vito” fue reconocida en 1872 por el médico norteamericano George Summer Huntington , quien hiciera la primera descripción clínica completa y clara de una enfermedad.

Enfermedad de Huntington
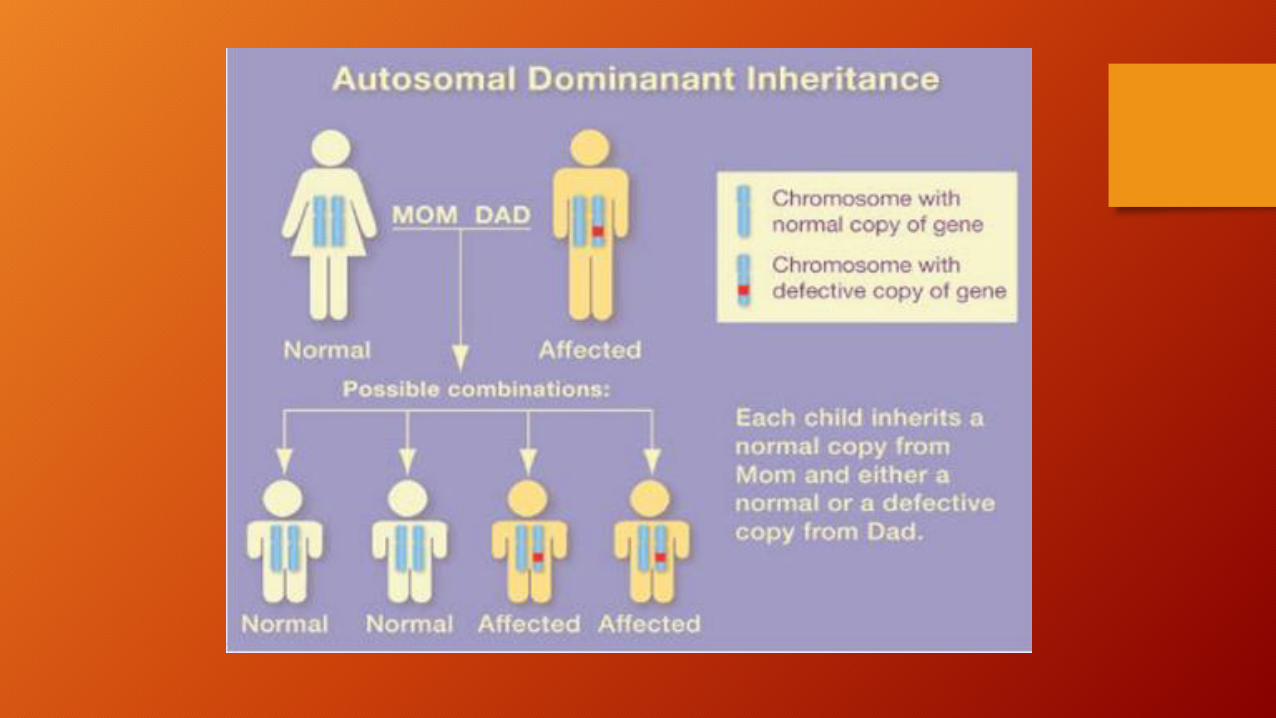
• La enfermedad de Huntington (EH) es un trastorno neurodegenerativo hereditario del sistema nervioso central que afecta principalmente a los ganglios basales.
• Se transmite de modo autosómico dominante
• Se manifiesta en el adulto a una edad variable:
• Típicamente entre los 30 y los 40 años de edad
• Formas juveniles (alrededor del 10%), de inicio antes de los 20 años
• Formas tardías (alrededor del 25%), después de los 50 años.

EPIDEMIOLOGIA
• La enfermedad está distribuida en todo el mundo en igual proporción entre hombres y mujeres.
• Prevalencia: se considera entre 5 y 10 casos por 100.000 habitantes, algo menor en países del este asiático y en la población de raza negra.
• Incidencia: anual varía entre 1 y 4 casos por millón de habitantes.
• Origen de la enfermedad: viene del oeste de Europa (Francia, Alemania y Holanda), con posterior dispersión hacia América, Inglaterra, Sudáfrica y Australia.
• Las mayores tasas de prevalencia: se dan en la región del lago de Maracaibo de Venezuela, en la isla de Tasmania (Sur de Australia) y en Moray Firth de Escocia .

Etiología
• La EH se trata de una de las 10 enfermedades hereditarias autosómicas dominantes, producidas por expansión excesiva de tripletes CAG (citosina-adenina-guanina) en sus respectivas proteínas (enfermedades poliglutamínicas o poliQ) :
Enfermedad Proteína
Enfermedad de
Huntington Huntingtina

Etiología
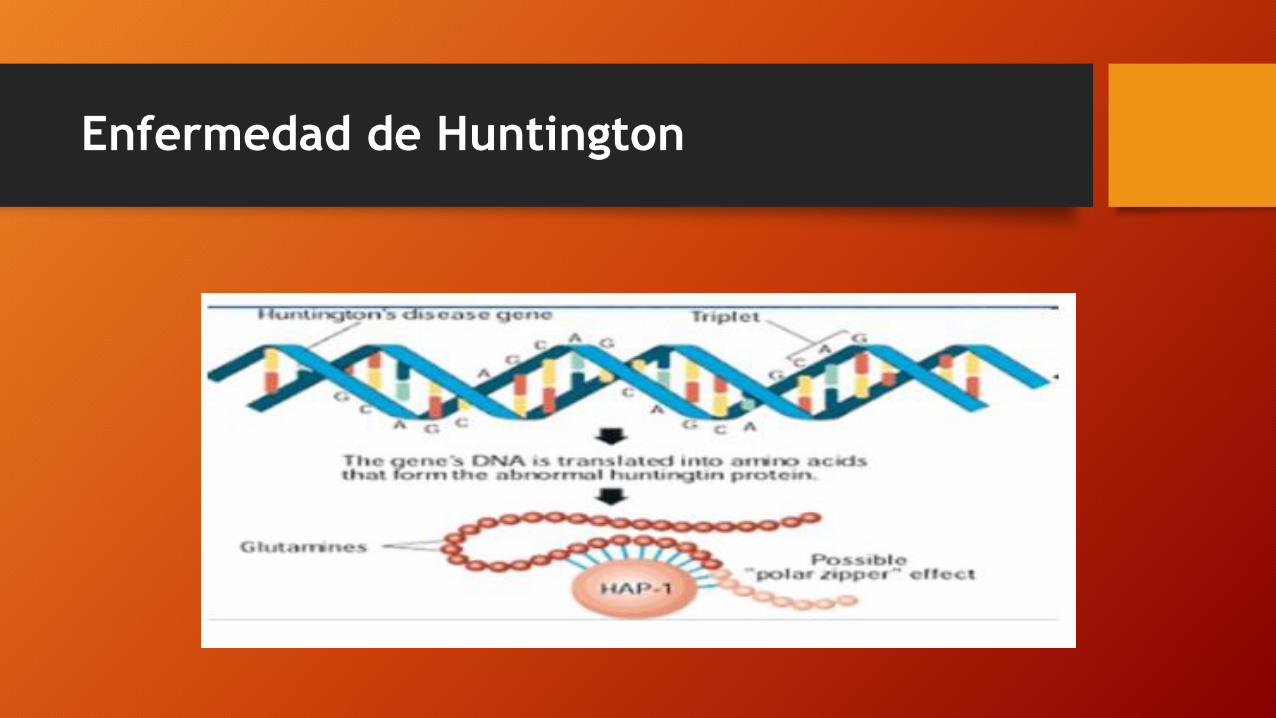
• Una mutación expansiva (CAG) en el primer exón del gen IT15 , situado en el cromosoma 4p16.3.
• Este gen se expande 210 kb y codifica para la huntingtina, una proteína de expresión ubicua en el núcleo o citoplasma de las células de diversos tejidos, incluidas las neuronas.
• La proteína mutante forma agregados nucleares, pero los mecanismos del proceso de neurodegeneración todavía hoy se desconocen.
• Expresión similar en heterocigotos y homocigotos
• Casos de inicio temprano usualmente tienen herencia paterna
Enfermedad de Huntington

Etiología
• El número de copias de este triplete en un individuo normal es menor de 35.
• De 36 a 39 repeticiones no tiene tanta penetrancia.
• Cuando hay 40 o más repeticiones, se produce la EH.
• Las expansiones entre 40 y 50 repeticiones de CAG son vistas con frecuencia en personas que presentan síntomas entre los 30 y 50 años.
• La EH juvenil (EHJ) se asocia con casos que sobrepasan las 70 repeticiones.



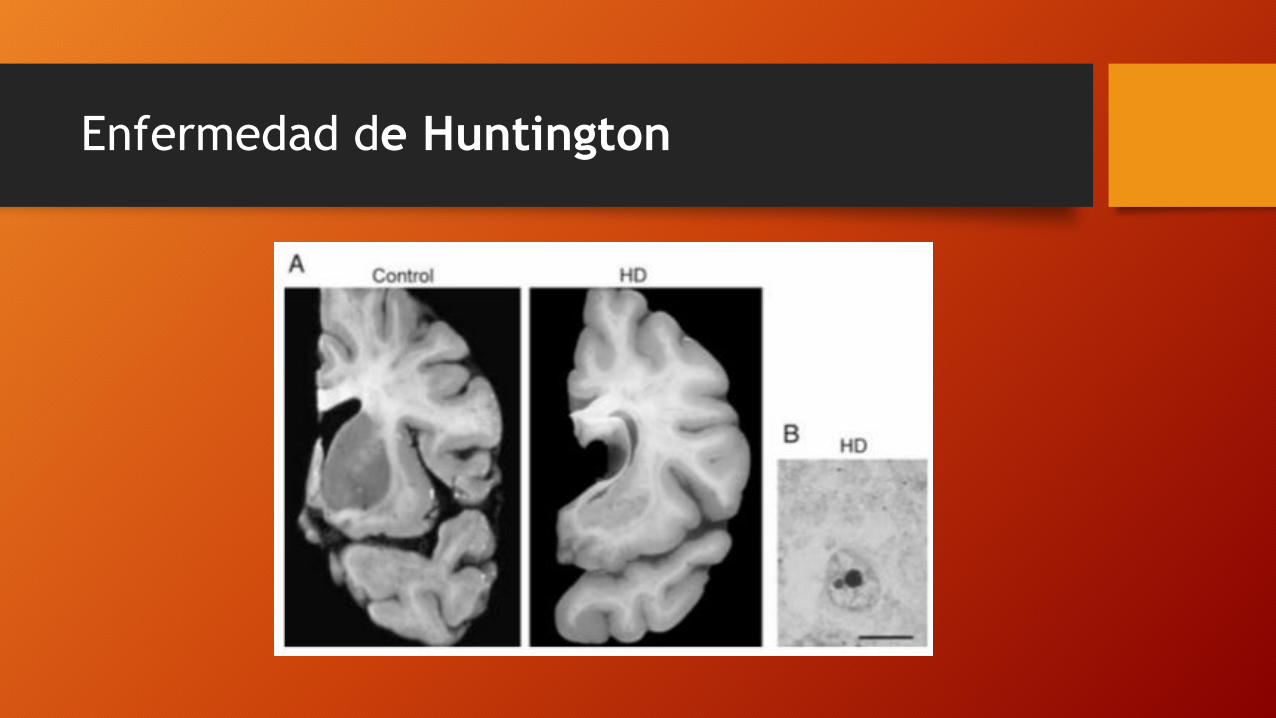
Características clínicas
• Demencia progresiva
•
• Movimientos coreicos
•
• Inicio característico en la quinta década
•
• Estos síntomas evolucionan hacia un empeoramiento progresivo y conducen, en un período de 10 a 20 años, a un síndrome demencial con un estado de postración en cama y caquexia

Signos y síntomas
• Los síntomas adicionales que pueden estar asociados con esta enfermedad son:
• Ansiedad
• Estrés
• Tensión
• Dificultad para deglutir
• Deterioro del habla
Enfermedad de Huntington

Pruebas diagnosticas
• El médico llevará a cabo un examen físico y puede hacer preguntas acerca de los síntomas y los antecedentes familiares del paciente.
• También se llevará a cabo un examen del sistema nervioso.
• El médico puede ver signos de:
• Demencia
• Movimientos anormales
• Reflejos anormales
• Marcha amplia y con "pavoneo"
• Lenguaje dubitativo o articulación deficiente

Pruebas de diagnostico
• Otros exámenes que pueden mostrar signos de enfermedad de Huntington son:
• Pruebas psicológicas
• TAC
• TEP (isótopos) del cerebro

Tratamiento
• El tratamiento debe ser multidisciplinario y aportar apoyo no solo al paciente sino también a sus familiares, por lo que es recomendable que intervengan asistentes sociales, genetistas, psicólogos y enfermeras conocedoras de la problemática de estos pacientes, además del neurólogo.

Asesoramiento Genético
• Acudir a una clínica genética permite disponer de información cierta y actualizada sobre la enfermedad.
• Habitualmente se mantiene una consulta con un genetista, que le permite comentar todas sus dudas referentes a la EH.
• Si decidiera pasar el test predictivo, tendría varias citas médicas con el equipo que le guiaría durante todo el proceso.
• se extraería una muestra de sangre la revelación de los resultados se realizaría a las 2-8 semanas
• La edad mínima recomendada para pasar el test predictivo son los 18 años, pues se supone que la persona tiene la madurez suficiente como para ser consciente de lo que significa ser portador del gen anómalo.
• En casos excepcionales, puede ser aconsejable realizar el test genético a niños que, por ejemplo, muestran signos de EH juvenil o en menores de 18 años si están embarazadas.

Asesoramiento Genético
• El análisis genético es una prueba de ADN que determina el número de repeticiones CAG del gen de Huntington y, por lo tanto, detecta la mutación de la EH.
• Se pueden diferenciar cuatro tipos de resultados:
• Por debajo de 27 repeticiones CAG, significa que se es una persona normal.
• Entre 27 y 35 repeticiones, significa que se es una persona normal pero con un pequeño riesgo de que el número aumente en generaciones futuras.
• Entre 36 y 39 repeticiones, el resultado es anormal, pero hay posibilidad de que la enfermedad se desarrolle a una edad muy avanzada o de que no llegue a hacerlo.
• Por encima de 40 repeticiones, el gen es anormal.

Catch 22

• Microdeleción 22q11
• Monosomía 22q11
• Síndrome DiGeorge
• Síndrome cardiofacial de Cayler
• Síndrome de Sedlackova
• Síndrome de Shprintzen
• Síndrome de Takao
• Síndrome de anomalías conotruncales y de la cara
• Síndrome velocardiofacial
•

Patrones de herencia
• La deleción del cromosoma 22 en la región q11 puede ser heredada, pero el 94 por ciento son deleciones “de novo”.

Cuadro clínico
• Malformaciones congénitas cuyos rasgos característicos incluyen:
• Defectos cardíacos
• Anomalías del paladar.
• Dimorfismo facial.
• Retraso en el desarrollo.
• inmunodeficiencia.

Incidencia
• 1 de cada 4000 nacimientos.
• 1 de cada 68 bebés con padecimientos cardiológicos congénitos y en un 5 a 8% de bebés con paladar hendido.

Diagnóstico
• Diagnóstico prenatal(En ultrasonido,una amniocentesis (extracción del líquido amniótico) para el estudio de cromosomas mediante “fluorescencia in situ hibridación” o FISH. Esta técnica utiliza una sonda de DNA de la región del síndrome en el cromosoma 22 y puede confirmar el 95% de los casos)
• Diagnóstico postnatal.
• Análisis de sangre y estudios para determinar problemas en el sistema inmunológico
• una placa de rayos X de tórax
• •Ecocardiografía

Tratamiento
•La edad del bebé, su estado general de salud y su historia médica. •Los signos y síntomas que presente. •La tolerancia que tenga el niño a determinados medicamentos, procedimientos o terapias.

Asesoría
• Se recomienda acudir al Consejo Genético ya que en la mayoría de los casos existe un defecto en los cromosomas y este desorden puede ser detectado durante el embarazo.

Sx. de DiGeorge
Sx.de DiGeorge
• etiología muy heterogénea se subdivide en 2 grandes grupos de pacientes:
• a) los pacientes portadores de la microdeleción 22q11.
• b) aquellos que podrían deberse a otras causas, en quienes un camino patogénico común, sería capaz de producir un fenotipo idéntico.


Sx.velocardiofacial
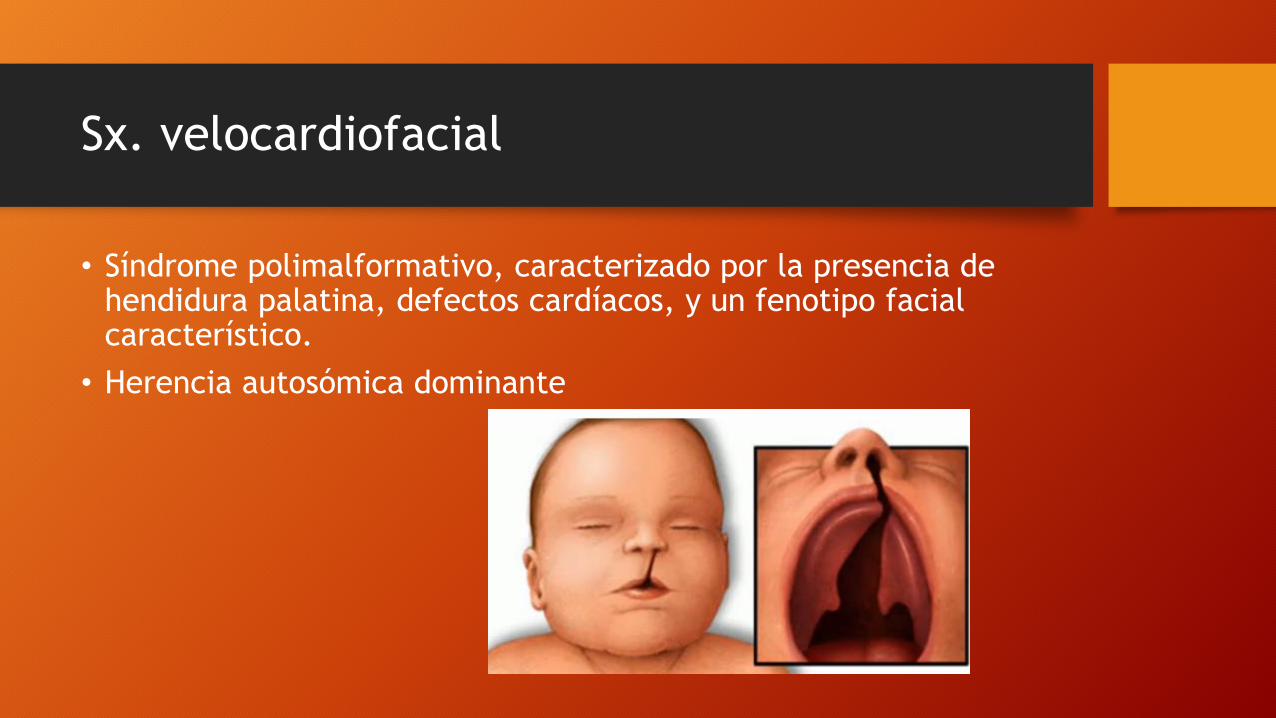
Sx. velocardiofacial
• Síndrome polimalformativo, caracterizado por la presencia de hendidura palatina, defectos cardíacos, y un fenotipo facial característico.
• Herencia autosómica dominante


Acondroplasia

Acondroplasia
• La causa de este trastorno es una mutación en el gen que codifica para el receptor 3 del factor de crecimiento de fibroblastos, localizado en el cromosoma 4

Etiología
• La herencia de este trastorno es autosómica dominante. Las posibilidades genotípicas y sus correspondientes fenotípicas son:
• Homocigoto
• Heterocigoto
• El 80% de los afectados no tienen antecedentes familiares. El motivo de estas mutaciones “De Novo” es que afectan a la línea germinal paterna

Signos y síntomas
• Muestran una presencia física como consecuencia de la interrupción del desarrollo del cartílago en las epífisis.
• Tienen una baja estatura, con acortamiento de extremidades y agrandamiento del cráneo.

Manejo
• Actualmente no se tiene un tratamiento para personas con acondroplasia
• La GH se usa para personas que no tienen acondroplasia, sin embargo no es efectiva en personas que si lo padecen
Epidemiología
• Las estimaciones de su prevalencia son difíciles puesto que los criterios de su diagnóstico son subjetivos y suelen cambiar con el tiempo.
• Afecta a 1 de cada 10.000 nacimientos

Marfan

Ehlers-Danlos
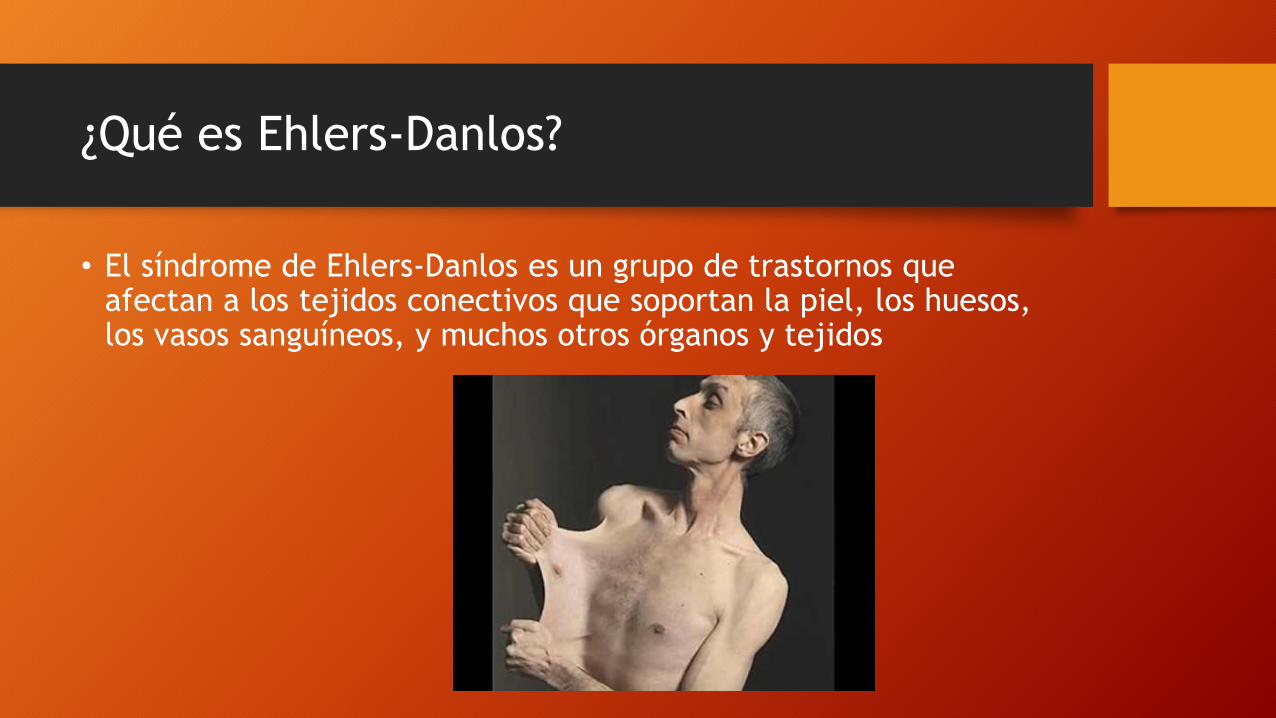
¿Qué es Ehlers-Danlos?
• El síndrome de Ehlers-Danlos es un grupo de trastornos que afectan a los tejidos conectivos que soportan la piel, los huesos, los vasos sanguíneos, y muchos otros órganos y tejidos

Tipos
• el tipo clásico
• el tipo de la hipermovilidad
• el tipo vascular
• el tipo cifoescoliosis
• el tipo Artrocalasia
• el tipo dermatosparaxis.

Cuadro Clínico
• Dolor de espalda
• Hiperlaxitud ligamentaria
• Piel que se estira, presenta equimosis y se daña fácilmente
• Cicatrización fácil y curación de heridas deficiente
• Pies planos
• Aumento de la movilidad articular, crujido en las articulaciones, artritis temprana
• Dislocación articular
• Dolor articular
• Ruptura prematura de membranas durante el parto
• Piel muy suave y aterciopelada
• Problemas de visión

Incidencia
• 1 de cada 5.000 personas en todo el mundo.
• Las formas clásicas hipermovilidad ;1 de cada 10.000 a 15.000 personas.
• el tipo clásico ;1 de cada 20.000 a 40.000 personas.
• El tipo vascular ;1 de cada 250.000 personas.

Mutaciones
• Los resultados tipo clásico ;gen COL5A1 o el gen COL5A2.
• Hipermovilidad;en el gen TNXB
• vasculares ;gen COL3A1.
• Cifoescoliosis;genes PLOD1.
• Artrocalasia;gen COL1A1 o COL1A2
• Dermatosparaxis; el gen ADAMTS2.

Patrón de herencia
• El patrón de herencia del síndrome de Ehlers-Danlos varía según el tipo.
• El Artrocalasia, clásica, hipermovilidad, y formas vasculares de la enfermedad tienen un patrón de herencia autosómico dominante.
• Otros casos son el resultado de nuevas mutaciones genéticas (esporádica).
• Los tipos dermatosparaxis y cifoescoliosis del síndrome de Ehlers-Danlos, se heredan con un patrón autosómico recesivo

Diagnóstico
• Tipificación del colágeno (realizada en una muestra de biopsia de piel).
• Prueba de mutación del gen de colágeno.
• Ecocardiografía (ecografía o ultrasonido del corazón).
• Actividad de lisil oxidasa o de lisil hidroxilasa (para verificar la formación de colágeno).

Tratamiento
• fisioterapia o la evaluación de un médico especialista en medicina de rehabilitación.

Asesoría
• En personas diagnosticadas de SED, es esencial el control médico de su caso destinado a identificar y prevenir los riesgos para su salud derivados de la enfermedad.
• se recomienda realizar una consulta de consejo genético a futuros padres que presenten la enfermedad o que conozcan la existencia de antecedentes de la misma en su familia, realizar las pruebas de análisis genético que se estimen necesarias.

Osteogenesis imperfecta

Osteogenesis imperfecta
• Es un grupo de trastornos hereditarios que predisponen a la fractura de los huesos incluso en traumatismos leves, así como a las deformidades esqueléticas.
• Se han reconocido una importante gama de variaciones clínicas

Etiología e incidencia
• Autosómica dominante
• Aproximadamente el 90% de los individuos afectados, presentan mutaciones en los dos genes (COL1A1 y COL1A2) que codifican las cadenas del colágeno tipo 1, la proteína principal del hueso.

Etiología e incidencia
• La heterogeneidad clínica se puede explicar, al menos en parte, por heterogeneidad de locus y por heterogeneidad alélica.
• Los fenotipos varían en función de la cadena de procolageno tipo I que esta afecta y según en tipo y la localización de la mutación en locus.

Etiología e incidencia
• La incidencia combinada de todas las formas de la enfermedad es aproximadamente de un paciente por cada 15,000 personas



Características clínicas
• escleróticas azules.
• tienen pérdida auditiva que comienza durante la segunda década de vida.
• Piel más rígida y menos elástica que la piel normal.
• prognatismo mandibular.
• Se rompen los huesos con facilidad.
• Articulaciones flexibles.
• Cara triangular.
• Dientes frágiles.

Diagnostico
• Clínica. Historia de fracturas, deformidades óseas, cifoescoliosis, retraso del crecimiento, escleras azules, dentinogénesis imperfecta, hipoacusia, antecedentes familiares.
• Hallazgos radiológicos. Lo más frecuente es la presencia de fracturas, osteopenia (disminución de densidad y contenido mineral óseo) variable en función de la gravedad clínica, rarefacción ósea progresiva, callos múltiples de distinta antigüedad, etc.

Neurofibromatosis-1
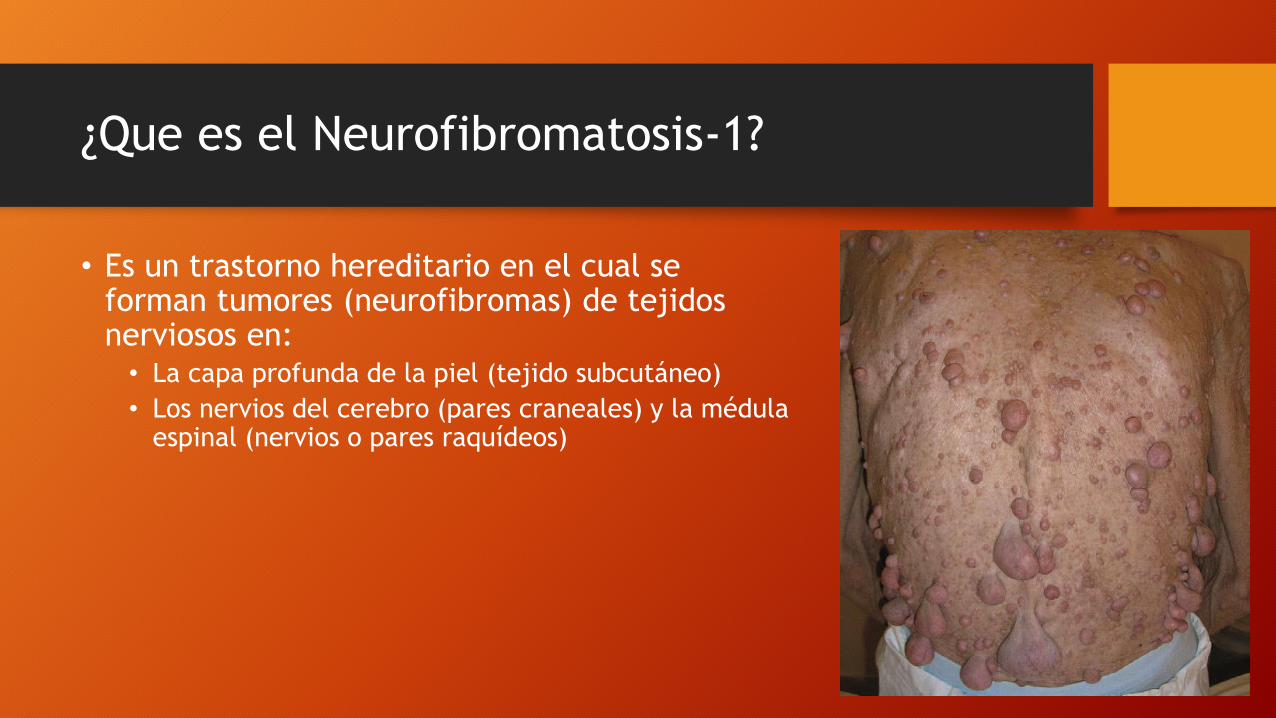
¿Que es el Neurofibromatosis-1?
• Es un trastorno hereditario en el cual se forman tumores (neurofibromas) de tejidos nerviosos en: • La capa profunda de la piel (tejido subcutáneo)
• Los nervios del cerebro (pares craneales) y la médula espinal (nervios o pares raquídeos)

• En las neurofibromatosis la herencia es autosómica dominante, lo que quiere decir que con tener un gen alterado de alguno de los padres aparecerá la enfermedad; así mismo, existe un riesgo de transmitir la enfermedad del 50 % a cada uno de los hijos.

Mutación e incidencia
• En las neurofibromatosis la herencia es autosómica dominante, lo que quiere decir que con tener un gen alterado de alguno de los padres aparecerá la enfermedad; existe un riesgo de transmitir la enfermedad del 50 % a cada uno de los hijos, o puede ocurrir que se deba a una mutación nueva.
• Una mutación es una alteración azarosa que puede ocurrir en el gen NF1 cuando el esperma o el óvulo se están formando. Las mutaciones espontáneas no están causadas por nada.
• 1 de cada 3000 nacidos, lo que indica una alta incidencia dentro de las enfermedades hereditarias.

Cuadro clínico
• La neurofibromatosis causa crecimiento incontrolado de tejido a lo largo de los nervios. • Causando dolor, daño nervioso
grave y perdida de la función en la zona estimulada por dicho nervio.
• Así como también presentar problemas de sensibilidad o el movimiento según nervio afectado.
• Las manchas de color café con leche son el síntoma distintivo de la neurofibromatosis. Adultos con 6 o más manchas superiores a 1.5 cm (0.5 cm en los niños) de diámetro tienen probabilidad de padecer neurofibromatosis.

Los signos que aparecen en esta categoría son:
• Corta Estatura - una proporción significativa tanto mientras son niños como cuando llegan a adultos. No se conoce la razón para esto y no existe una anormalidad relacionada que pueda explicar este hecho.
• Macrocefalia (cabeza más grande) - las personas con NF1 tienden a tener cabezas un poco más grandes que la población en general.

Manejo
• Se recomienda la asesoría genética para cualquier persona con antecedentes familiares de neurofibromatosis.
• Asimismo, se recomienda de manera enfática hacerse exámenes anuales de los ojos, la piel, la espalda, el sistema nervioso (neurológicos) y vigilancia de la presión arterial.

Asesoramiento genético
• Consulte con el médico si:
• Observa manchas de color café con leche en la piel de su hijo o cualquier otro síntoma de esta afección.
• Tiene antecedentes familiares de neurofibromatosis y está planeando tener hijos o le gustaría que examinaran a su hijo.

Tratamiento
• No existe un tratamiento específico para la neurofibromatosis. • Los tumores que causan dolor o pérdida de alguna función se pueden
extirpar.
• Los tumores que han crecido de manera rápida se deben extirpar inmediatamente puesto que pueden tornarse cancerosos (malignos).
• Algunos niños con trastornos de aprendizaje pueden necesitar enseñanza especial.

Síndrome de esclerosis tuberosa