Dermatomiositis juvenil
48
Dermatomiositis Juvenil Revisión de tema MR2 Ramon Florez CEMENA 2012
-
Upload
ramon-florez -
Category
Health & Medicine
-
view
2.603 -
download
5
Transcript of Dermatomiositis juvenil

Dermatomiositis Juvenil
Revisión de tema
MR2 Ramon FlorezCEMENA 2012

Introducción
Definición
La dermatomiositis juvenil y la polimiositisjuvenil son raras, frecuentemente
crónicas, miopatías autoinmunes de la infancia.

Reumatología 2005; 21(1):33-35

Epidemiología• Miopatía inflamatoria idiopática: – 85% DMJ– 2 – 8% PMJ
• Incidencia: 2-3 /1.000.000 en USA al año• Inicio promedio 7 años . 25 % antes de los 4
años.Peak: 5 – 10 años• Más frecuente en sexo femenino: 2-3:1 USA v/s
5:1 en UK.Rheumatology (Oxford). 2006 Oct;45(10):1255-60. Epub 2006 Mar 27.
Rheum Dis Clin North Am. 2002 Nov;28(4):833-57.
Juvenile dermatomyositis and other idiopathic infl ammatory myopathies of childhoodLancet 2008; 371: 2201–12

Patogénesis
• Etiología poco clara• Reacción autoinmune en el musculo estriado
de individuos genéticamente susceptibles, posiblemente en respuesta a desencadenantes ambientales.

Susceptibilidad genética
• Mayor prevalencia en gemelos monocigotos y familiares de primer grado.
• Mayor prevalencia de algunos HLA (B8, DQA1*0501, DQA1*0301).
• Polimorfismo genético del TNF-alfa.
Arthritis Rheum. 2006 Dec;54(12):3979-87.J Rheumatol. 1998 May;25(5):1000-2.

Mecanismos inmunológicos
• Invasión del tejido muscular por linfocitos T CD8+ en la PMJ.
• AAN (+) en un 70%• Auto anticuerpos específicos de miositis (Anti
Jo-1, Anti Mi-2)• Microquimerismo materno
Rheum Dis Clin North Am 1994 Nov;20(4):919-42.Lancet 2000 Dec 23-30;356(9248):2155-6.Lancet 2000 Dec 23-30;356(9248):2156-7.
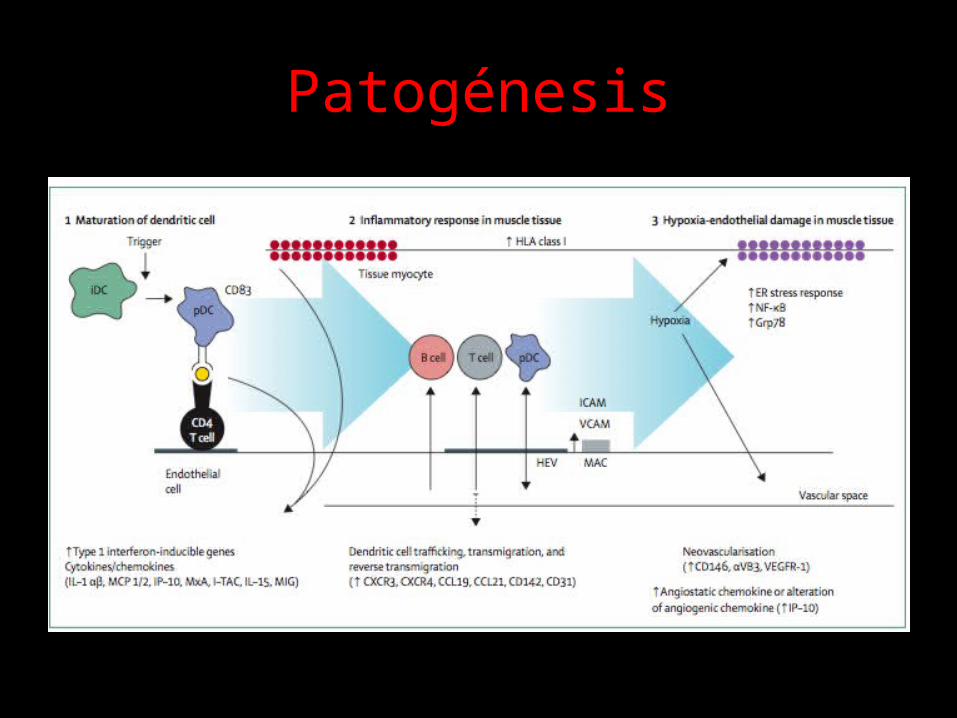
Patogénesis

Curr Op Rheumatol 2004; 16:692-699.

Factores ambientales
• La exposición a radiación ultravioleta estimularía la producción de auto anticuerpos Mi-2
Curr Op Rheumatol 2004; 16:700-706.

Infección
• Respuesta inusual a una infección en huésped susceptible.
• Coxsackie B, echovirus, Epstein Barr, CMV, s. pyogenes, toxoplasma, enterovirus, parvovirus.

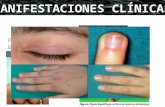
Manifestaciones Clínicas
• La debilidad muscular proximal constituye el principal síntoma.
• Rash característico en DMJ• Otros síntomas (cefalea, fiebre, pérdida de
peso, fatiga).

• Estudio en centro terciario de Canada, 105 pacientes con DMJ (edad media al dg 7,6 años).
– Pápulas de Gottron: 91%– Rash heliotropo: 83%– Rash malar/facial: 42%– Cambios de lecho capilar ungueal: 80%– Mialgias/artralgias: 25%– Disfonía o disfagia: 24%– Anorexia: 18%– Fiebre: 16%
Manifestaciones Clínicas
Rheum Dis Clin North Am. 2002 Nov;28(4):833-57.

Manifestaciones cutáneas
• Rash: – Rash heliotropo (dermatitis heliotropa): rash rojo
–violáceo del párpado superior, acompañado de edema. Asociado a eritema malar/facial
– Pápulas de Gottron: erupción eritematosa, papuloescamosa, en la superficie dorsal de los nudillos. Lesiones similares en superficies extensoras (codos, rodilla, maleolo interno)



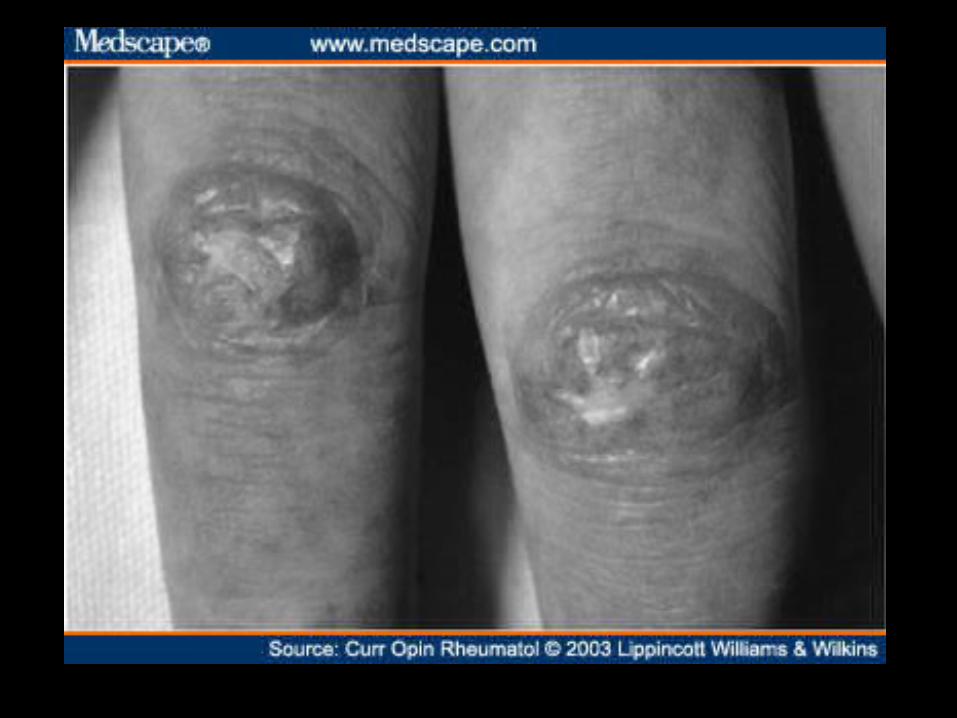

Cambios en lecho capilar ungueal
• Dilatación capilar, tortuosidad y disminución del número de capilares.

Capilaroscopía del lecho ungueal
• Se coloca una gota de aceite en la base ungueal y se mira con microscopía directa o con un oftalmoscopio.
• Su alteración refleja enfermedad del tejido conectivo.
• Útil como apoyo diagnóstico y para el seguimiento.

Capilaroscopía del lecho ungueal
Rev.Colomb.Reumatol. vol.15 no.3 Bogotá July/Sep. 2008


Ulceraciones cutáneas
• Manifestación seria• Refleja mayor severidad y peor pronóstico de
la DMJ.


Calcinosis• Calcificación de partes blandas.• Se desarrolla más tarde en la historia natural
de la enfermedad (entre 1 a 3 años de evolución; se ha descrito hasta 20 años).
• Amplia presentación clínica (nódulos, placas superficiales, calcificaciones musculares tumorales, difusas en la fascia muscular, etc).
• Pueden provocar dolor y limitar la función articular.



Dermatomiositis amiopática
• Presentación sin debilidad muscular. • Muy infrecuente.

Debilidad muscular
• Simétrica• Proximal (cintura pélvica y escapular)• Generalmente de inicio en miembros inferiores• Generalmente comienzo insidioso• Limitación funcional (subir escaleras, pararse del suelo,
subirse a un automóvil).• El signo de Gowers suele ser positivo.• Debilidad del paladar blando, m. cricofaríngeo
(deglución).• Compromiso del 1/3 superior del esófago (disfagia)

Signo de Gowers

Otros síntomas
• Artritis• Lipodistrofia• Enfermedad pulmonar intersticial (adultos)• Vasculopatía gastrointestinal (complicación),
puede llevar a la perforación intestinal.


Criterios DiagnósticosClasificación de Bohan y Peter (1975)

Criterios diagnósticos
• Debilidad proximal simétrica• Rash característico– Dermatitis heliotropa– Pápulas de Gottron
• Elevación de enzimas musculares• Común elevación de transaminasas; GOT y GPT.• Electromiografía sugerente, patrón característico (miopatía y
denervación en reposo y en esfuerzo aparecen potenciales polifásicos de breve duración).
• Biopsia muscular : Patognomónico de DM es la atrofia perifascicular.

Diagnóstico
• Principalmente clínico• EMG y pruebas de laboratorio apoyan el
diagnóstico.• La biopsia muscular a pesar de ser un examen
invasivo se recomienda realizar en los pacientes con clínica y laboratorio sugerente de DMJ, ya que existen signos patognomónicos en ella.

Exámenes complementarios
• Enzimas musculares– Se encuentran elevadas (hasta 50 veces el valor
normal en 2/3 de los pacientes) e indican daño muscular.
– Se solicitan: Creatinkinasa (CK), Lactato deshidrogenasa (LDH), ALT (GPT), AST (GOT).
– Pueden encontrarse normales en un 15 a 25% de los casos, por lo que enzimas normales no descartan la enfermedad.
J Pediatr. 2006 Feb;148(2):247-253.

Exámenes complementarios
• Electromiografía• Resonancia nuclear magnética de músculos• Biopsia muscular• ANA, Anti Jo-1, Anti Mi-2
J Pediatr. 2006 Feb;148(2):247-253.

Diagnóstico Diferencial
• En ausencia del característico compromiso cutáneo.• Miopatías no inflamatorias: Distrofia muscular de
Duchenne y Becker (pacientes hombres)• Miopatías metabólicas: déficit de carnitina, defectos
mitocondriales.• Miopatías infecciosas: influenza, coxsackie,
poliomielitis.• Otras reumatológicas: escleroderma, LES, enfermedad
mixta del tejido conectivo, ARJ.

Tratamiento• Metas: – Control de la miositis inflamatoria– Prevención y tratamiento de las complicaciones
• Tratamiento inicial dependerá de la presentación clínica.
• Patrones de la enfermedad:– Monocíclica– Policíclica– Persistente crónica
• Tratamiento precoz

Terapia inmunosupresora
• El manejo está basado en estudios observacionales y experiencia clínica.
• No existen ensayos clínicos controlados aleatorizados.
• En general, se inicia la terapia con una combinación de corticoides y metotrexato.

Glucocorticoides
• Agente inicial de elección• Disminuye mortalidad y complicaciones– Prednisona 2mg/kg/día (dosis máxima 80mg/día)– Metilprednisolona en pulsos 30mg/kg/día (máx
1g) por 3 días. Luego semanal o mensual (o prednisona).
• Efectos adversos de uso prolongado: retardo del crecimiento, aspecto cushingoideo, hipertensión arterial, cataratas, osteoporosis.

Agentes secundarios• Beneficio de uso como terapia inicial v/s
respuesta incompleta a corticoides.• Metotrexato es el fármaco más utilizado. – Acorta el tiempo de uso de corticoides y reduce su
dosis acumulativa.– 15mg/m2/semana (máx. 25mg/dosis)– Náuseas y vómitos, elevación de transaminasas,
infecciones.– Aportar ácido fólico 1mg/día

Agentes secundarios• Inmunoglobulina EV– 2g/kg ev (máx. 70g)– En pacientes con resistencia o dependencia a
corticoides.• Ciclosporina– En DMJ refractaria– 3 – 5mg/kg/día
• Ciclofosfamida– Reservado para casos severos y que amenazan la
vida.

Agentes secundarios• Agentes biológicos:– Rituximab– Infliximab
• Otros:– Micofenolato mofetil– Tacrolimus– Hidroxicloroquina

Otras terapias
• Protección solar• Corticoides/tacrolimus tópico• Fisioterapia y terapia ocupacional– Mejorar fuerza muscular, capacidad aeróbica y
movimiento articular.

Complicaciones
• Osteoporosis– Calcio 1000mg/día + Vitamina D 400 UI– Bifosfonatos: alendronato
• Calcinosis– Asociada a manejo tardío y/o inadecuado
• Perforación intestinal– Rara. Se ve en presentaciones severas

Pronóstico
• Mortalidad en los años ‘60: 30%• Con terapia inmunosupresora: 2-3%• Curso monocíclico ocurre aprox. En 1/3 de los
pacientes. Buena evolución.• Curso policíclico y persistente: 2/3– Riesgo de dolor persistente, calcinosis, limitación
funcional.

Muchas gracias