
UNIVERSITE PARIS VAL-DE-MARNE FACULTE DE MEDECINE DE …doxa.u-pec.fr/theses/th0216841.pdf · IV-3....
89
UNIVERSITE PARIS VAL-DE-MARNE FACULTE DE MEDECINE DE CRETEIL ANNEE : 2004 DES N° MEMOIRE DU DIPLOME D’ETUDES SPECIALISEES DE BIOLOGIE MEDICALE Conformément aux dispositions de l’Arrêté du 10 septembre 1990 tient lieu de THESE Pour l’obtention du Diplôme d’Etat de DOCTEUR EN MEDECINE Présentée et soutenue publiquement devant le Jury Interrégional le 25 octobre 2004 Par Elise CHAPIRO Née le 4 Novembre 1976 à Corbeil-Essonnes EXPRESSION DE TRANSCRITS AFFILIES A LA LIGNEE LYMPHOIDE T ET PRESENCE DE REARRANGEMENTS DES TCR DANS LA LEUCEMIE AIGUE PROMYELOCYTAIRE IMPLICATIONS POUR LA CIBLE CELLULAIRE DE LA t(15 ;17) JURY Président : Professeur Elizabeth Macintyre Membres : Docteur Vahid Asnafi Professeur Hélène Merle-Béral Professeur Michel Arock Professeur Christine Chomienne PRESIDENT DE THESE : LE CONSERVATEUR DE LA Professeur Elizabeth Macintyre BIBLIOTHEQUE UNIVERSITAIRE DIRECTEUR DE THESE Docteur Vahid Asnafi Signature du Président de thèse Cachet de la Bibliothèque Universitaire
Transcript of UNIVERSITE PARIS VAL-DE-MARNE FACULTE DE MEDECINE DE …doxa.u-pec.fr/theses/th0216841.pdf · IV-3....

UNIVERSITE PARIS VAL-DE-MARNE
FACULTE DE MEDECINE DE CRETEIL
ANNEE : 2004 DES N°
MEMOIRE DU DIPLOME D’ETUDES SPECIALISEES
DE BIOLOGIE MEDICALE
Conformément aux dispositions de l’Arrêté du 10 septembre 1990 tient lieu de
THESE
Pour l’obtention du Diplôme d’Etat de
DOCTEUR EN MEDECINE
Présentée et soutenue publiquement devant le Jury Interrégional le 25 octobre 2004
Par Elise CHAPIRO Née le 4 Novembre 1976 à Corbeil-Essonnes
EXPRESSION DE TRANSCRITS AFFILIES A LA LIGNEE LYMPHOIDE T ET PRESENCE DE REARRANGEMENTS DES TCR DANS LA LEUCEMIE AIGUE
PROMYELOCYTAIRE
IMPLICATIONS POUR LA CIBLE CELLULAIRE DE LA t(15 ;17)
JURY Président : Professeur Elizabeth Macintyre Membres : Docteur Vahid Asnafi
Professeur Hélène Merle-Béral Professeur Michel Arock Professeur Christine Chomienne PRESIDENT DE THESE : LE CONSERVATEUR DE LA Professeur Elizabeth Macintyre BIBLIOTHEQUE UNIVERSITAIRE DIRECTEUR DE THESE Docteur Vahid Asnafi Signature du Président de thèse Cachet de la Bibliothèque Universitaire

2
REMERCIEMENTS
A Madame le Professeur E. Macintyre,
Vous m'avez fait l'honneur d'accepter de présider cette thèse,
Pour m’avoir permis de réaliser ce travail passionnant et pour votre soutien,
Veuillez trouver ici l’expression de mes plus vifs remerciements et de mon profond respect.
A Monsieur le Docteur V. Asnafi,
Tu m'as fait l'honneur d'accepter de diriger cette thèse,
Pour ta grande disponibilité, tes précieux conseils, tes remarques pertinentes, et ta bonne
humeur,
Trouve ici le témoignage de ma très sincère reconnaissance et tous mes remerciements.
A Madame le Professeur H. Merle-Béral,
Vous avez accepté d’être membre du jury de cette thèse,
Pour votre soutien depuis mes débuts en hématologie,
Veuillez trouver ici l'expression de mon profond respect ainsi que ma très grande gratitude
A Monsieur le Professeur M. Arock,
Vous m'avez fait l'honneur d'accepter de juger cette thèse,
Veuillez recevoir ma très respectueuse considération.
A Madame le Professeur C. Chomienne,
Vous avez accepté de faire partie du jury de cette thèse,
Veuillez recevoir mes très sincères remerciements.
A l'équipe du laboratoire d'Hématologie de l'Hôpital Necker-Enfants Malades
Veuillez trouver ici mes plus grands remerciements pour votre aide et pour le temps que vous
m'avez accordé afin que ce travail puisse aboutir.
Un grand merci à Corinne Millien qui m’a enseigné avec patience, gentillesse et disponibilité
les techniques de biologie moléculaire.
Merci à Patrick, Daniel, Maryse, Catherine pour leur aide technique et informatique.

3
A Monsieur le Professeur David Grimwade,
Pour votre précieuse collaboration,
Veuillez recevoir mes sincères remerciements.
A mes parents,
Pour votre soutien permanent, votre générosité, votre confiance, votre éducation exemplaire
qui m’ont permis de réaliser tous mes projets,
Veuillez trouver ici le témoignage de toute la reconnaissance et de tout l’amour que je vous
porte.
A ma sœur, Lucile,
Merci pour ton écoute et ton aide précieuse en anglais.
Avec toute mon affection.
A Fabrice,
Merci infiniment de m’avoir accompagné pendant toutes ces années d’études avec tendresse et
patience, de m’avoir toujours soutenue et réconfortée dans les périodes de doutes, et d’avoir
partagé avec moi les moments de bonheur.
A mes amis (Barbara, Julie-Gaëlle, Magali-Anne, Greg, Julie, Seb, Jérôme, Nico, Isabelle,
Anne-Cécile, Ivan…),
Pour vos encouragements et les agréables moments de détente passés ensemble.
Avec toute mon amitié.

4
SOMMAIRE
INTRODUCTION............................................................................................. 10
I. LA LEUCEMIE AIGUE PROMYELOCYTAIRE......................................................... 10
I-1. Généralités sur les leucémies aiguës myéloblastiques............................................................................ 10 I-1-1. Epidémiologie ..................................................................................................................................... 10 I-1-2. Diagnostic des LAM ........................................................................................................................... 11
I-2. Généralités sur la leucémie aiguë promyélocytaire ............................................................................... 15 I-3. Pathogenèse moléculaire de la leucémie aiguë promyélocytaire .......................................................... 17
I-3-1. Variants moléculaires de PML-RARα ................................................................................................ 19 I-3-2. RARα et la granulopoièse normale ..................................................................................................... 19 I-3-3. Rôle de PML ....................................................................................................................................... 20 I-3-4. Mécanismes de pathogenèse de PML-RARα ...................................................................................... 21 I-3-5. La protéine PML-RARα est-elle suffisante pour induire une LAP ? .................................................. 22
I-4. Nature de la cellule cible de la t(15 ;17).................................................................................................. 23 I-4-1. Le concept de la cellule souche leucémique........................................................................................ 23 I-4-2. Arguments en faveur de la survenue de la t(15 ;17) dans un progéniteur engagé dans la lignée myéloïde........................................................................................................................................................ 24 I-4-3. Arguments en faveur de la survenue de la t(15 ;17) dans un progéniteur primitif............................... 25
II. HEMATOPOIESE ET DIFFERENCIATION LYMPHOCYTAIRE T PRECOCE. 27
II-1. Les modèles de l’hématopoïèse .............................................................................................................. 27 II-2. Le thymus : organe de la lymphopoïèse T............................................................................................. 31 II-3. Etapes précoces de la lymphopoïèse T .................................................................................................. 32 II-4. ββββ-sélection................................................................................................................................................ 34
II-4-1. Le Pré-TCR........................................................................................................................................ 34 II-4-2. Pré-Tα (pTα) ..................................................................................................................................... 35
II-5. Réarrangement des loci du TCR ........................................................................................................... 36 II-5-1. Définitions ......................................................................................................................................... 36 II-5-2. Organisation des gènes du TCR......................................................................................................... 37
II-5-2-1. Locus δ ...................................................................................................................................... 37 II-5-2-2. Locus γ ....................................................................................................................................... 38
II-5-3. Mécanisme de la recombinaison V(D)J et rôle de RAG .................................................................... 39 II-5-3-1. Processus de recombinaison ...................................................................................................... 39 II-5-3-2. Expression des protéines RAG1 et RAG2................................................................................... 41 II-5-3-3. Régulation de la recombinaison V(D)J ...................................................................................... 41
II-5-4. Cinétique de réarrangement des loci du TCR au cours de la maturation thymique............................ 42 II-5-5. Rôle de TEA ...................................................................................................................................... 42
IV. OBJECTIFS DU TRAVAIL ........................................................................................... 43
MATERIEL ET METHODES......................................................................... 45
I. PATIENTS .......................................................................................................................... 45
II. PREPARATION DES CELLULES ................................................................................ 45
III. IMMUNOPHENOTYPAGE .......................................................................................... 46

5
III.1. Principe de la cytométrie en flux .......................................................................................................... 46 III-2. Marquage des cellules ........................................................................................................................... 46 III-3. Analyse de l’expression des marqueurs............................................................................................... 47
IV. QUANTIFICATION DES TRANSCRITS PAR RQ-PCR .......................................... 48
IV-1. Extraction des ARN totaux................................................................................................................... 48 IV-2. Transcription Inverse............................................................................................................................ 48 IV-3. PCR quantitative en temps réel TaqMan® .......................................................................................... 49
IV-3-1. Principe ............................................................................................................................................ 49 IV-3-2. Conditions de la PCR ....................................................................................................................... 49 IV-3-3. Amorces et sondes............................................................................................................................ 51 IV-3-4. Quantification et expression des résultats......................................................................................... 51
V. REARRANGEMENTS DES TCR δδδδ et γγγγ ......................................................................... 52
V-1. Principe .................................................................................................................................................... 52 V-2. Extraction d’ADN ................................................................................................................................... 52 V-3. Réarrangements du TCR δδδδ .................................................................................................................... 53
V-3-1. PCR multiplexe.................................................................................................................................. 53 V-3-1-1. Conditions de PCR et séquence des amorces ............................................................................. 53 V-3-1-2. Analyse des produits d’amplification ......................................................................................... 55
V-3-2. PCR triplex ........................................................................................................................................ 55 V-3-2-1. Conditions de PCR et séquence des amorces ............................................................................. 55 V-3-2-2. Analyse des produits d’amplification ......................................................................................... 55
V-4. Réarrangements du TCRγγγγ ...................................................................................................................... 56 V-4.1. Conditions de PCR et séquence des amorces..................................................................................... 56 V-4-2. Analyse des produits d’amplification................................................................................................. 56
VI. RECHERCHE D’UNE DUPLICATION EN TANDEM DE FLT3 (FLT3-ITD) ...... 58
VI-1. Conditions de la PCR............................................................................................................................ 58 VI-2. Séquences des oligonucléotides............................................................................................................. 58 VI-3. Analyse des produits d’amplification................................................................................................... 58
VII. STATISTIQUES ............................................................................................................ 60
RESULTATS ..................................................................................................... 61
I. CARACTERISATION DES ECHANTILLONS.......................................................... 61
II. EXPRESSION DE TRANSCRITS LYMPHOÏDES T PRECOCES DANS LA LAP 62
II-1. Les blastes de LAP expriment pTαααα mais pas RAG1 ............................................................................ 62 II-1-1. pTα .................................................................................................................................................... 62 II-1-2. RAG1................................................................................................................................................. 63
II-2. Les blastes de LAP expriment TEA....................................................................................................... 64 II-3. Expression de TdT et CD3 cytoplasmique............................................................................................ 66
III. REARRANGEMENTS DES TCR δδδδ et γγγγ ....................................................................... 67
IV. EXPRESSION DE pTα α α α ET TEA EN FONCTION DES SOUS-GROUPES de LAP68

6
DISCUSSION..................................................................................................... 69
I. INFIDELITE DE LIGNEE................................................................................................ 69
I-1. Action directe de PML-RARαααα sur l’expression des gènes affiliés à la lignée T .................................. 69 I-2. Instabilité génomique et dérégulation secondaire de l’expression des gènes....................................... 70
I-2-1. Nécessité d’évènements secondaires pour aboutir au phénotype leucémique complet........................ 70 I-2-2. Quels mécanismes pourraient être à l’origine de l’acquisition d’évènements secondaires ? ............... 71
I-3. Limites du modèle de l’infidélité de lignée ............................................................................................. 72
II. PROMISCUITE DE LIGNEE ......................................................................................... 72
II-1. Légitimité du modèle de la promiscuité de lignée................................................................................. 73 II-2. Comment replacer la cellule cible de la t(15 ;17) dans les LAPv dans l’hématopoïèse à la lumière des marqueurs T retrouvés ?.......................................................................................................................... 74
II-2-1. Transformation d’un progéniteur avant l’engagement vers la lignée myéloïde.................................. 74 II-2-2. Quel progéniteur non engagé définitivement dans la lignée myéloïde pourrait être la cible de la t(15 ;17) ?...................................................................................................................................................... 75 II-2-3. LAP microgranulaire et LAP classique: origine cellulaire différente ?.............................................. 75
II-3. Interprétation hypothétique des niveaux de pTαααα/TEA dans les LAM contrôles .............................. 77 II-4. Limites du modèle de la promiscuité de lignée ..................................................................................... 77
III. CONCLUSION ................................................................................................................ 78
CONCLUSION ET PERSPECTIVES ............................................................ 80
BIBLIOGRAPHIE............................................................................................ 81

7
LISTE DES ABBREVIATIONS
Abl : gène Abelson
Ac : anticorps
ADN : Acide désoxyribonucléique
ADNc : Acide désoxyribonucléique complémentaire
Ag : antigène
AR : acide rétinoïque
ARN : Acide ribonucléique
ARNm : Acide ribonucléique messager
ATRA : all trans retinoic acid
BCR : breaking cluster region
BFU-E : burst forming unit-erythoid
CD : cluster de différenciation
CFU : colony forming unit
CFU-G : colony forming unit-Granular
CFU-GM : colony forming unit-Granular Monocytic
CSH : cellule souche hématopoïétique
CIVD : coagulation intra-vasculaire disséminée
CLP : common lymphoid progenitor
CMLP : common myeloid lymphoid progenitor
CMP : common myeloid progenitor
CMH : complexe majeur d’histocompatiblité
CSL : cellule souche leucémique
Ct : cycle threshold
DN : double négatif
dNTP : déoxynucléotides tri phosphate
DMSO : diméthyl sulfoxyde
DO : densité optique
DP : double positif

8
EDP : early double positif
EDTA : éthylène diamide tétracétique
FAB : French American British
FISH : fluorescence in situ hybridation
FLT3 : Fms-like tyrosine kinase 3
FLT3-ITD : FLT3 internal tandem duplication
hCG : cathepsine G humaine
hMRP8 : macrophage-inhibiting factor related protein 8 humaine
HDAC : histone dé-acétylase
HPC : hematopoietic progenitor cell
ISP : immature simple positif
Ig : immunoglobuline
IgH : chaîne lourde d’immunoglobuline
IL7R : interleukine 7 receptor
LA : leucémie aiguë
LAL : leucémie aiguë lymphoblastique
LAM : leucémie aiguë myéloblastique
LAP : leucémie aiguë promyélocytaire
LAPv : leucémie aiguë promyélocytaire de morphologie variante, microgranulaire
LAPc : leucémie aiguë promyélocytaire de morphologie classique, hypergranulaire
MPO : myélopéroxydase
MLL : mixed lineage leukemia
NB : nuclear body
NK : natural killer
NOD/SCID : non obese diabetic/severe combinated immunodeficient
Pb : paire de bases
PBS : Phosphate-buffered saline
PCR : polymerase chain reaction
pTα : pré-T alpha
PML : promyelocytic leukemia
RAG 1 et 2 : recombinase activating gene 1 et 2
RARα : retinoic acid receptor alpha
RARE : retinoic acid response element

9
RPMI : Roswell Park Memorial Institute (milieu de culture)
RT-PCR : reverse transcriptase - polymerase chain reaction
RSS : Recombinaison Signal Séquence
SL-IC : SCID leukemia-initiating cell
SP : simple positif
TAMRA : 6-carboxy-tétraméthyl-rhodamine
TCR : T cell receptor
TdT : déoxynucléotidyl terminale transférase
TEA : T early alpha

10
INTRODUCTION
I. LA LEUCEMIE AIGUE PROMYELOCYTAIRE
I-1. Généralités sur les leucémies aiguës myéloblastiques
I-1-1. Epidémiologie
Les leucémies aiguës (LA) sont des affections malignes développées au dépend des
cellules hématopoïétiques immatures. Ces hémopathies sont caractérisées par une
prolifération clonale de précurseurs médullaires bloqués à un stade précoce de leur maturation
(blastes). Cette prolifération siège initialement dans la moelle osseuse puis envahit le sang et
certains organes entraînant la constitution d’un syndrome tumoral (hépatosplénomégalie,
adénopathies...). Il s’y associe un défaut d’hématopoïèse aboutissant, à des degrés variables, à
la constitution de cytopénies : anémie, thrombopénie, neutropénie. Les complications liées à
l’insuffisance de production médullaire sont à l’origine de l’évolution toujours fatale de cette
pathologie en l’absence de traitement.
Classiquement, deux types de leucémies aiguës sont distinguées en fonction de
l’appartenance des blastes à la lignée myéloïde (leucémies aiguës myéloblastiques, LAM) ou
lymphoïde (leucémies aiguës lymphoblastiques, LAL).
La fréquence des leucémies aiguës est d’environ deux nouveaux cas pour 100 000
habitants par an, l’incidence augmentant avec l’âge. Les LAL représentent 80% des LA de
l’enfant et 20% des LA de l’adulte. Les LAM sont donc beaucoup plus fréquentes chez
l’adulte avec un âge médian de survenue de 60 ans. La fréquence est égale dans les deux
sexes.
Si la majorité des LA surviennent de novo (85%), sans étiologie retrouvée, certaines sont
secondaires à une exposition professionnelle au benzène ou à ses dérivés, à un traitement
antérieur par chimiothérapie (alkylants, étoposide, anthacyclines …) ou par radiations
ionisantes, ou à une hémopathie préexistante (syndrome myéloprolifératif ou

11
myélodysplasique).
I-1-2. Diagnostic des LAM
Le diagnostic des LAM repose sur un ensemble d’arguments à la fois cliniques,
morphologiques, phénotypiques, moléculaires et cytogénétiques.
Les signes cliniques surviennent en général de manière brutale. Ils sont en rapport soit
avec le syndrome tumoral, rare dans les LAM (hypertrophie gingivale, atteinte neuroméningée
adénopathies, surtout présentes dans les LAM monoblastiques), soit avec l’insuffisance
médullaire : anémie (pâleur, asthénie, dyspnée, tachycardie), neutropénie (fièvre isolée,
infections récidivantes), thrombopénie (hémorragies muqueuses, purpura pétéchial).
Le diagnostic et le classement des LA reposent sur l’examen cytologique des frottis de
sang et de moelle complété parfois par des réactions cytochimiques et/ou un phénotypage
immunologique. Le bilan comprend aussi une étude cytogénétique et moléculaire permettant
de préciser le type exact de la LA et son pronostic.
L’hémogramme n’est quasiment jamais normal. L’anémie et la thrombopénie sont
fréquentes. Le nombre de globules blancs est variable : normal, abaissé avec neutropénie, ou
élevé avec cellules blastiques circulantes. Le myélogramme par ponction sternale ou iliaque
objective une infiltration médullaire de plus de 20% de cellules blastiques (selon l’OMS).
L’analyse cytologique complétée si besoin par des études cytochimiques (réaction des
myélopéroxydases et des estérases) permet la classification FAB (French-American-British
Cooperative Group) [1] (tableau 1). Le phénotype immunologique est devenu un complément
permettant de confirmer et d’affiner le diagnostic. Il est moins essentiel que pour le diagnostic
des LAL mais permet de classer une LA peu différenciée.
L’étude cytogénétique conventionnelle et par hybridation in situ fluorescente (FISH)
permet de mettre en évidence des anomalies chromosomiques dans environ 50% des cas. La
détection d’anomalies chromosomiques contribue largement à préciser le pronostic des LAM
[2]. Trois groupes sont distingués en fonction des anomalies retrouvées. La présence d’une
translocation t(8 ;21), t(15 ;17), t(16 ;16) ou d’une inversion du chromosome 16 est de
pronostic favorable. Le groupe à risque intermédiaire correspond aux patients ayant un
caryotype normal ou certaines anomalies de nombre : +8, +21, +22, del(7q), del(9q) ou un
réarrangement du 11q23 impliquant le gène MLL (mixed lineage leukemia). Le groupe de

12
risque élevé concerne les cas avec caryotype complexe ou présentant une délétion du 5, du 7
ou un 3q anormal.
Les techniques de biologie moléculaire visent à détecter les transcrits de fusion
résultant des translocations chromosomiques récurrentes (AML1-ETO et t(8 ;21), PML-
RARα et t(15 ;17), CBFβ-MYH et inv16…). Leur intérêt essentiel est l’évaluation de la
maladie résiduelle avec une grande sensibilité.
Une nouvelle classification des LAM a été proposée en 1999 par l’organisation
mondiale de la santé (OMS). Cette classification, constituant désormais la classification de
référence, permet de tenir compte des facteurs pronostiques puisqu’elle intègre les données
cytogénétiques [3] (tableau 2).
Au total, les LAM forment un groupe hétérogène, tant au niveau de leur présentation
clinique que de leurs caractéristiques phénotypiques, moléculaires et cytogénétiques. Ainsi, la
caractérisation précise de chaque cas est indispensable pour apprécier le pronostic et adapter
au mieux les choix thérapeutiques.

13
Tableau 1. Classification FAB des LAM
Type FAB Caractéristiques cytologiques Cytochimie
LAM0 (très peu
différenciée)
Pas d’identité morphologique typique MPO et estérases -
LAM1 (sans maturation) Absence de maturation, quelques
granulations azurophiles
MPO +
LAM2 (avec maturation) Maturation granuleuse présente,
granulations fréquentes, corps d’Auer +
MPO+
Estérase +, non
inhibées par NaF
LAM3 (promyélocytaire) Nombreuses granulations
Corps d’Auer en fagots
Variante : microgranulaire, avec noyaux
bilobés
MPO+++
Estérase +, non
inhibées par NaF
LAM4
(myélomonocytaire)
Aspect de M2 avec >20% de monocytes
sanguins ou médullaires
Variante : LAM4 avec contingent
éosinophile
MPO +
Estérase+++, inhibées
par NaF
LAM5 (monoblastique) Monoblastes et promonocytes > 80% MPO +/-
Estérase+, inhibées
par NaF
LAM6 (érythroleucémie) Hyperplasie érythroblastique
Myéloblastes >30%
MPO et estérases -
LAM7
(mégacaryoblastique)
mégacaryoblastes MPO et estérases -

14
LAM avec translocations chromosomiques récurrentes
LAM avec t(8;21)(q22;q22), AML1 (CBF-alpha)/ETO
LAM3 avec t(15;17)(q22;q11-12), PML/RAR-alpha et variantes
LAM avec éosinophiles anormaux, inv(16) (p13q22) ou t(16;16)(p13;q22), CBF-
bêta/MYH11
LAM avec anomalies 11q23 (MLL)
LAM avec myélodysplasie « multilignée »
Avec antécédent de SMD
Sans antécédent de SMD
LAM et SMD « secondaires » à des thérapeutiques
Après agents alkylants
Après épidophyllotoxin
« secondaires » d’autres types
LAM n’entrant pas dans les catégories précédentes (retour à la classification FAB)
LAM avec différenciation minimale (M0)
LAM sans maturation (M1)
LAM avec maturation (M2)
LAM promyélocytaire (LAM3)
LAM avec différenciation myélomonocytaire (LAM4)
LAM monocytaire (LAM5)
LAM avec différenciation érythroblastique (M6)
LAM avec différenciation mégacaryocytaire (M7)
LAM avec différenciation basophile
LAM avec myélofibrose
LA biphénotypique
Tableau 2. Classification OMS des LAM

15
I-2. Généralités sur la leucémie aiguë promyélocytaire
La leucémie aiguë promyélocytaire (LAP) est un sous-type de leucémie aiguë
myéloblastique (LAM) représentant environ 10% des cas [4]. Elle se caractérise par
l’accumulation de cellules myéloïdes bloquées au stade promyélocytaire dans la moelle
osseuse et le sang périphérique. Cette pathologie survient chez des adultes relativement jeunes
(âge médian de 43 ans) avec une distribution égale dans les deux sexes.
Elle s’accompagne de manière quasi-constante d’une coagulation intra-vasculaire
disséminée (CIVD) pouvant provoquer des thromboses ou plus souvent des complications
hémorragiques risquant de mettre en jeu le pronostic vital, ce qui rend nécessaire un
diagnostic rapide. Cette coagulopathie était responsable de la mort d’environ un quart des
patients dans les anciennes séries. Actuellement, l’initiation précoce d’un traitement par
l’acide tout trans rétinoique (ATRA) permet de contrôler la CIVD et réduit la mortalité à
moins de 10% [5].
Sur le plan morphologique, la classification FAB distingue deux sous-types : la forme
classique hypergranulaire (LAPc) et la forme variante microgranulaire (LAPv) [1]. La forme
classique, fréquemment pancytopénique, se caractérise par des blastes hypergranuleux
contenant souvent des corps d’Auer en fagots. Dans la forme variante, habituellement
hyperleucocytaire, les blastes sont au contraire hypogranuleux, rarement avec corps d’Auer,
mais ont typiquement un noyau bilobé (figure 1). Les promyélocytes leucémiques présentent
une réaction myélopéroxidasique très forte dans les deux formes.
Les études immunophénotypiques révèlent un profil d’expression particulier avec une
forte positivité du CD33 et du CD13 associée à l’absence d’expression de HLA-DR et à la
faible fréquence du CD34 [6]. L’expression de CD2, marqueur associé à la lignée lymphoïde
T, est retrouvé dans environ 25% des cas [7-9]. Il existe une corrélation entre la morphologie
variante et l’expression du CD34 et du CD2 [9, 10]. Le marqueur spécifique de la lignée
lymphoïde B, CD19, est retrouvé dans environ 10% des cas, sans association à un sous-type
morphologique particulier [9].

16
Figure 1. Les deux types morphologiques de la leucémie aiguë promyélocytaire
En haut : la forme classique, hypergranulaire, avec corps d’Auer en fagot En bas : la forme variante, microgranulaire, avec noyaux bilobés

17
Depuis sa découverte il y a 25 ans, la translocation t(15 ;17)(q22 ;q21), aboutissant à la
production d’une protéine de fusion PML-RARα, a été considérée comme le marqueur
moléculaire des LAP. Cependant, 8% des cas ne présentent pas cette translocation. Dans
lamajorité de ces cas, l’analyse moléculaire révèle la présence du gène de fusion PML-RARα
résultant d’insertions pouvant être cryptiques cytogénétiquement (4%) ou de réarrangements
complexes (2%) [11]. Dans les autres cas (2%), des réarrangements du 17q21 conduisent à la
fusion de RARα avec des gènes partenaires alternatifs : PLZF (promyelocytic leukemia zinc
finger), NPM (nucleophosmin), NuMA (nuclear mitotic apparatus) et STAT5b associés aux
translocations t(11 ;17)(q23 ;q21) [12], t(5 ;17)(q35 ;q21) [13], t(11 ;17)(q13 ;q21) [14] et
der(17) [15], respectivement.
La LAP est actuellement considérée comme l’une des formes les plus favorables de
LAM. Ce bon pronostic est lié à l’utilisation d’un traitement par l’acide tout-trans-rétinoique
(all trans retinoic acid, ATRA) qui permet de cibler directement la lésion moléculaire et
aboutit au déblocage du processus de différenciation des cellules blastiques. L’administration
d’ATRA en association aux chimiothérapies d’induction (daunorubicine ou idarubicine)
permet d’atteindre des taux de rémission complète proches de 90% [16].
I-3. Pathogenèse moléculaire de la leucémie aiguë promyélocytaire
En 1977, Rowley et collègues ont identifié la translocation réciproque équilibrée
t(15 ;17) comme le marqueur caryotypique de la LAP [17] (figure 2), et c’est au début des
années 1990 qu’a été découverte l’implication de la fusion entre le gène RARα sur le
chromosome 17 et le gène initialement inconnu nommé PML (promyelocytic leukemia) sur le
chromosome 15 [18]. Depuis, d’autres translocations impliquant toutes RARα ont été
décrites, indiquant que l’anomalie de RARα est à l’origine de la pathogenèse de la LAP . La
t(15 ;17) étant retrouvée dans la très grande majorité des cas, nous décrirons ici les
mécanismes oncogénétiques aboutissant à la survenue d’une LAP liés à cette translocation [9,
19].

18
Figure 2. Translocation t(15 ;17)
Figure 2. Translocation t(15 ;17)
Figure 3. Variants moléculaires du transcrit de fusion PML-RARαααα

19
I-3-1. Variants moléculaires de PML-RARαααα
Trois formes moléculaires sont distinguées en fonction de la localisation du point de
cassure dans le gène PML sur le chromosome 15 (figure 3). Si le point de cassure survient au
niveau du sixième intron (55% des cas), le transcrit de fusion sera du type BCR1 (breaking
cluster region 1). Le type BCR2, plus rare (5%), est défini par une cassure localisée au sein du
6ème exon. Ces 2 types, de taille très proche, ne sont pas distingués actuellement par les
techniques de biologie moléculaire utilisées en routine. Le type BCR3, retrouvé dans environ
40% des cas et associé à la forme morphologique variante [9] correspond à un transcrit de
fusion plus court, avec un point de cassure plus en 5’ du gène PML, au niveau du 3ème intron.
Ces trois formes moléculaires aboutissent toutes à la formation d’un transcrit de fusion et
d’une protéine chimérique PML-RARα. Le point de cassure sur le chromosome 17 est unique,
au niveau du 2ème intron du gène RARα.
I-3-2. RARαααα et la granulopoièse normale
L’acide rétinoïque (AR) et son récepteur α (RARα) jouent un rôle important dans le
développement de la lignée myéloïde. Bien que cette voie ne soit pas cruciale dans la
granulopoièse, comme le montre les données issues des souris knock-out pour le gène RARα
qui présentent un taux de polynucléaires normal, elle joue cependant un rôle dans la
modulation/régulation de ce processus [20].
L’AR est un métabolite de la vitamine A dont l’activité biologique est médiée par
liaison à des récepteurs nucléaires spécifiques, les RARs (α, β et γ) et les RXRs (α, β et γ).
Ces deux types de récepteurs sont activés par l’acide rétinoïque 9-cis, RAR étant aussi
sensible à l’ATRA. Ces récepteurs forment des hétérodimères (RAR/RXR) se liant à des
motifs d’ADN nommés RAREs (retinoïc acid-response elements) localisés dans la région
promotrice des gènes cibles de la voie de l’AR.
En l’absence de ligand, ces hétérodimères sont capables de lier les séquences RAREs
avec une forte affinité et exercent une activité répressive sur la transcription des gènes en
s’associant à un complexe contenant des co-répresseurs N-CoR (nuclear receptor co-
repressor). Ces co-répresseurs, à leur tour, recrutent des histone-déacétylases (HDAC),

20
entraînant une déacétylation des histones et une condensation de la chromatine. La
chromatine, dans cette configuration, est inaccessible à la machinerie transcriptionnelle, il en
résulte donc une répression des gènes cibles.
La liaison de l’AR à son récepteur RARα induit un changement conformationnel de
RARα permettant la dissociation du complexe co-répresseur et le recrutement de molécules
co-activatrices (CBP, ACTR…) qui entraînent une acétylation des histones et donc une
décondensation de la chromatine. Ce relâchement chromatinien permet au complexe
d’initiation de la transcription d’accéder à l’ADN et donc d’activer l’expression des gènes
nécessaires à la différenciation granuleuse.
RARα a donc une fonction double : non lié, il apparaît comme un régulateur négatif de
la maturation myéloïde alors que lié, il stimule cette différenciation.
I-3-3. Rôle de PML
Le gène PML, exprimé de façon ubiquitaire, code pour une protéine localisée au sein
de domaines nucléaires nommés « corps nucléaires » (PML-nuclear bodies, NBs), ND10
(nuclear domain 10) ou PODs (PML oncogénic domains) [21]. Ces NBs sont au nombre de
10 à 30 par noyau. Ils sont composés de plus de trente protéines ayant des fonctions cellulaires
diverses. PML est essentiel à la formation de ces NBs comme le montrent la désorganisation
des corps nucléaires dans les cellules PML-/- et en présence de la protéine de fusion PML-
RARα [22].
Les fonctions précises de PML ne sont pas encore entièrement claires. L’étude des
souris knock-out PML-/- et de son hyperexpression dans des lignées cellulaires d’origine
diverses a permis d’impliquer PML dans plusieurs processus cellulaires incluant l’apoptose, la
croissance cellulaire, la suppression de tumeur, la régulation transcriptionnelle, la stabilité
génomique et la réponse aux infections virales [23]. PML a un rôle de suppresseur de tumeur
comme l’indique la susceptibilité accrue des souris PML-/- au développement de tumeurs
après exposition à des carcinogènes [24]. Cette activité s’exerce à plusieurs niveaux, par
contrôle de la croissance cellulaire, de la survie cellulaire et de la stabilité génomique.
L’hyperexpression de PML induit un arrêt de croissance de toutes les lignées testées, et les
cellules PML-/- en culture ont une croissance plus élevée que les cellules normales, indiquant
un rôle d’inhibition du cycle cellulaire [25]. PML est également un facteur pro-apoptotique

21
impliqué dans les voies dépendante et indépendante de p53 et a été impliqué dans le maintien
de la stabilité génomique.
I-3-4. Mécanismes de pathogenèse de PML-RARαααα
La t(15 ;17) génère 2 protéines de fusion PML-RARα et RARα-PML, mais c’est
l’isoforme PML-RARα qui contribue majoritairement à la pathogenèse de la LAP. Cette
protéine de fusion conserve les domaines principaux de chacune des 2 protéines : la portion
RARα reste capable de lier l’ADN et de s’hétérodimériser avec RXR. Cependant,
contrairement à RARα, PML-RARα peut également lier les séquences RAREs sous forme
d’homodimères. En conséquence, la liaison des homodimères PML-RARα et des
hétérodimères PML-RARα/RXR aux séquences RAREs, a un effet dominant-négatif sur la
protéine RARα sauvage. En l’absence de ligand, PML-RARα réprime la transcription plus
fortement que RARα sauvage car elle s’associe aux co-répresseurs de façon plus étroite, et
des taux physiologiques d’AR (10-9-10-8 M) ne sont pas capables de les dissocier. De plus, la
répression transcriptionnelle est accrue par une méthylation aberrante de l’ADN, liée au
recrutement d’ADN méthyltransférases par la protéine de fusion [26]. D’autre part, la
formation d’hétérodimères PML-RARα/RXR provoque une séquestration de RXR qui ne peut
interférer avec d’autres récepteurs nucléaires d’hormone comme le récepteur aux hormones
thyroïdiennes qui requiert l’association aux RXR pour lier l’ADN. Enfin, PML-RARα se lie à
PML, ce qui entraîne la fragmentation des corps nucléaires et donc la délocalisation de PML
et des autres composants des NBs.
Le traitement par l’ATRA à des concentrations pharmacologiques (10-7-10-6M) agit
selon deux modes d’action : premièrement, il induit une dissociation des co-répresseurs,
conduisant à l’activation des gènes cibles et donc à la poursuite de la différenciation des
promyélocytes. Deuxièmement, l’ATRA induit la dégradation de PML-RARα qui devient
incapable de lier PML, ce qui permet la reformation des corps nucléaires. De plus, cette
dégradation induit le relargage de RXR qui est alors libre d’interagir avec la protéine RARα
sauvage et les autres récepteurs d’hormone nucléaires.

22
I-3-5. La protéine PML-RARαααα est-elle suffisante pour induire une LAP ?
Les études sur lignées cellulaires et les modèles in vivo ont déterminé l’importance de
la protéine de fusion PML-RARα dans la pathogenèse des LAP. L’expression de PML-RARα
dans la lignée promonocytaire U937 lui fait perdre sa capacité de différenciation à différents
stimuli et entraîne une croissance cellulaire accrue liée à une inhibition de l’apoptose [27]. La
transduction de progéniteurs hématopoïétiques humains purifiés (HPC) par des vecteurs
rétroviraux codant pour PML-RARα induit le développement d’un phénotype de LAP. Une
différenciation rapide en promyélocytes suivie d’un arrêt de maturation aboli par l’AR sont
observés dans ce modèle, ainsi qu’un engagement préférentiel dans la différenciation
granuleuse indépendant de tout stimulus par un facteur de croissance hématopoïétique. Ces
observations montrent que PML-RARα est directement impliqué dans le stade de
différenciation des cellules leucémiques [28].
Cependant, l’étude des modèles de souris transgéniques montre que PML-RARα est
nécessaire mais insuffisant pour le développement d’une LAP. Grisolano et collègues [29] ont
utilisé des souris exprimant PML-RARα sous le contrôle de séquences régulatrices du gène de
la cathepsine G humaine (hCG) exprimée dans les promyélocytes. Ces souris transgéniques
présentent une altération de la maturation granuleuse (accumulation de précurseurs myéloïdes)
mais seulement 30% d’entre-elles développent une LAP et ce, après une longue période de
latence. De même, la granulopoièse est altérée chez les souris exprimant PML-RARα sous le
contrôle du promoteur de la hMRP8 (macrophage-inhibiting factor related protein 8
humaine) [30] ou CD11b [31] mais celles-ci ne développent une LAP que rarement (10-30%)
et après plusieurs mois d’évolution (6-12 mois). La pénétrance incomplète ainsi que la longue
période de latence observées dans ces souris transgéniques PML-RARα suggèrent que PML-
RARα est clairement requis mais n’est pas suffisant pour provoquer une LAP : des
évènements génétiques additionnels doivent survenir. Un modèle murin récent de LAP basé
sur la transduction par un vecteur rétroviral de progéniteurs hématopoïétiques murins lin-
purifiés renforce l’hypothèse de la nécessité de deux évènements oncogéniques [32]. En effet,
ces cellules montrent des capacités prolifératives augmentées et des anomalies de
différenciation terminale in vitro. L’inoculation de ces cellules dans des souris syngéniques
irradiées provoque le développement de LAP sensibles à l’ATRA avec une forte fréquence
(>80%) et une courte période de latence (4 mois). L’absence d’anomalie évidente à l’état

23
préleucémique, ajoutée à la monoclonalité ou l’oligoclonalité des blastes leucémiques chez
ces souris est en faveur de la nécessité de lésions génétiques additionnelles. La pénétrance
accrue du phénotype leucémique dans les souris transgéniques en présence du produit de
fusion réciproque RARα-PML [33], de duplication de FLT3 (FLT3-ITD) [34] ou de
l’hyperexpression de bcl2 [35] conforte cette hypothèse.
I-4. Nature de la cellule cible de la t(15 ;17)
I-4-1. Le concept de la cellule souche leucémique
Le concept de la cellule souche leucémique a émergé dans les années 1970 en
s’appuyant sur plusieurs études montrant que seule une petite sous-population de cellules
leucémiques avaient des capacités de prolifération intense. Par exemple, quand des cellules
myélomateuses murines issues d’ascite de souris sont séparées des cellules souches
hématopoïétiques normales puis cultivées in vitro, seulement 1 sur 10000 à 1 sur 1000
cellules tumorales sont capables de former des colonies [36]. De plus, seulement 1 à 4% des
cellules leucémiques greffées in vivo peuvent former des colonies spléniques quel que soit le
type de leucémie [37]. Cette capacité proliférative limitée des blastes leucémiques suggère
que seule une petite population de cellules souches leucémiques présente le potentiel d’auto-
renouvellement nécessaire à la pérennisation du clone leucémique. L’étude de Bonnet et Dick
[38] est la claire démonstration de l’existence des cellules souches leucémiques (CSL). Dans
ces travaux, des souris NOD/SCID (non obese diabetic/severe combined immunodeficient)
irradiées sont greffées avec des blastes totaux ou triés en fonction de l’expression du CD34 et
du CD38 issus de patients atteints de différents types de LAM, puis l’envahissement de la
moelle osseuse par les cellules humaines est évalué par Southern Blot. Les résultats montrent
que seule la fraction CD34++CD38-, représentant une faible proportion des cellules blastiques
totales (0.2 à 1%) peut conférer le phénotype leucémique aux souris NOD/SCID. Ces cellules
sont dénommées SL-IC (SCID leukemia-initiating cell). Elles sont capables de générer un
grand nombre de cellules blastiques exprimant les mêmes aberrations phénotypiques que
celles présentes initialement chez le patient, indiquant que le clone est organisé de façon
hiérarchique comme l’hématopoïèse normale : les cellules souches dotées d’un fort potentiel
prolifératif et d’auto-renouvellement donnent naissance de façon clonale aux cellules

24
constituant la majorité des cellules tumorales, peu proliférantes. Celles-ci ne gardent pas
toujours les caractères fonctionnels et morphologiques de la cellule souche si cette dernière
conserve la possibilité de se différencier partiellement.
Deux hypothèses peuvent être proposées concernant l’origine des cellules souches
leucémiques [39]. Le fait que les cellules souches normales (CSH) et les CSL partagent les
mêmes capacités d’auto-renouvellement amène à penser que les CSL sont des CSH devenues
leucémiques par l’accumulation de mutations. Les cellules souches ont une machinerie
d’auto-renouvellement déjà active pouvant être plus facilement dérégulée que dans une cellule
plus différenciée. De plus, les cellules souches ont une durée de vie longue qui facilite
l’accumulation de mutations. A l’opposé, les CSL pourraient être des progéniteurs plus
différenciés ou même des cellules matures qui auraient réacquis les capacités
d’autorenouvellement des cellules souches avant de devenir tumorale par accumulation de
mutations additionnelles.
Les expériences menées par Bonnet et Dick [38] sont en faveur de la première
hypothèse. Pour tous les sous-types de LAM (sauf les LAP), les seules cellules capables de
coloniser la moelle des souris NOD/SCID ont un phénotype CD34++CD38- similaire à celui
des cellules souches normales reconstituant l’hématopoïèse des souris SCID. Ces données
suggèrent que les cellules primitives normales, plus que les progéniteurs engagés, sont la cible
de la transformation leucémique dans les LAM. Cependant, dans ce travail, les cellules issues
de 3 échantillons de LAP sont incapables de coloniser la moelle des souris NOD/SCID
contrairement aux autres types de LAM. Cette observation suggère que la LAP est une entité
distincte dans laquelle le processus leucémique impliquerait un progéniteur engagé dans la
lignée myéloïde.
I-4-2. Arguments en faveur de la survenue de la t(15 ;17) dans un progéniteur
engagé dans la lignée myéloïde
D’autres arguments viennent appuyer cette hypothèse. Notamment, l’analyse de la
clonalité et de l’expression de PML-RARα dans des sous-populations médullaires purifiées
par tri cellulaire provenant de 3 cas de LAP a montré que seule la fraction CD34+CD38+
contenait le transcrit de fusion. Ces données suggèrent que la cellule souche primitive
(CD34+CD38-) n’est pas impliquée dans le processus néoplasique de la LAP [40].
Une autre étude, employant une analyse morphologique et une technique FICTION,

25
combinant analyses par FISH et immunophénotypiques aboutit aux mêmes conclusions [41].
Dans chacun des 6 cas étudié (LAPc), la fusion PML-RARα est restreinte à la lignée
myéloïde, sans positivité retrouvée au niveau des érythroblastes, des plasmocytes ou des
lymphocytes B et T.
De plus, les modèles murins de LAP ont été établis en utilisant des promoteurs qui
sont activés relativement tard dans le développement myéloïde (MRP8, Cathepsine G) alors
que l’expression de PML-RARα à un niveau élevé sous le contrôle du promoteur de la β-
actine, ubiquitaire, induit une létalité embryonnaire.
Une approche expérimentale alternative de modélisation mathématique basé sur les
données épidémiologiques du registre britannique des leucémies dont le but est de définir le
nombre d’étapes oncogéniques nécessaires au développement d’une LAP, prédit qu’un
progéniteur engagé dans la différenciation myéloïde est la cible d’un événement leucémogène
unique [42].
I-4-3. Arguments en faveur de la survenue de la t(15 ;17) dans un progéniteur
primitif
Alors que plusieurs arguments renforcent l’hypothèse d’une transformation au niveau
d’un progéniteur engagé dans la lignée myéloïde, il est difficile de replacer certaines données
expérimentales dans ce contexte.
En effet, Edwards et col. ont démontré la présence de la fusion PML-RARα par FISH
dans la majorité des cellules de la fraction CD34+CD38- dans 2 cas de LAP microgranulaires
[43].
Une analyse de 5 échantillons de LAP (de forme morphologique non précisée) par RT-PCR a
montré la présence de PML-RARα dans les CFU-GM et les BFU-E dans 2 cas, suggérant que
la t(15 ;17) survient dans une cellule souche pluripotente chez certains patients [44].
D’autre part, l’étude immunophénotypique de la LAP a révélé certains profils
d’expression atypiques pour un progéniteur engagé. Certains cas expriment CD34 (25%
environ), le marqueur affilié à la lignée NK CD56 (15%), le marqueur lymphocytaire B CD19
(15%) et le marqueur spécifique de la lignée T CD2 (25% des cas). Il a été confirmé que la
positivité du CD2 en surface était le reflet du niveau d’expression de l’ARNm qui est
comparable à celui détecté dans les lymphocytes T [8, 45].

26
Le mécanisme de cette expression aberrante de marqueurs lymphoïdes reste encore obscur et
est sujet à débat depuis plus de 20 ans. Selon le modèle de l’« infidélité de lignée », ce
phénomène est la conséquence de la dérégulation des gènes liée au processus leucémique,
tandis que selon le modèle de la « promiscuité de lignée », la co-expression de marqueurs
myéloïdes et lymphoïdes est le reflet de l’immunophénotype de la cellule cible de la
transformation leucémique qui est perpétué dans la descendance du clone malin. Afin de
comprendre les mécanismes inhérents à cette co-expression, une étude récente a analysé la
conformation chromatinienne au niveau du locus CD2 par un test d’hypersensibilité à l’
ADNase I dans les blastes de LAP [45]. La majorité des cas étaient des formes
microgranulaires. Chaque cas présentait une conformation chromatinienne ouverte semblable
à celle retrouvée dans les lymphocytes T, quel que soit le statut du CD2 à la surface
membranaire. Or, des études ont suggéré que, dans les progéniteurs hématopoïétiques
précoces, certains gènes considérés comme restreints à une lignée, incluant la β-globine, IgH,
MPO, sont en conformation chromatinienne ouverte avant l’engagement vers une lignée
particulière [46]. De plus, des analyses en RT-PCR sur cellule unique ont établi que des
transcrits de multiples lignées étaient co-exprimés dans les progéniteurs multipotents dérivés
de moelle osseuse humaine ou de souris [47]. La conformation chromatinienne retrouvée au
niveau du locus CD2 dans la LAP pourrait donc être le reflet de celle du progéniteur cible de
la t(15 ;17), suggérant ainsi que la LAP survient dans un progéniteur immature, non encore
engagé dans la lignée myéloïde, ayant un potentiel T.
Afin de comprendre dans quelle mesure les cellules blastiques de LAP sont
apparentées à la lignée lymphoïde T, nous avons étudié l’expression de transcrits T-
spécifiques précoces (pTα, TEA et RAG1) et recherché des réarrangements des TCR δ et γ.
Nous allons maintenant détailler la lymphopoïèse T, exposer le rôle de ces transcrits dans ce
processus et décrire les réarrangements des TCR après avoir rappelé quelques notions sur
l’hématopoïèse.

27
II. HEMATOPOIESE ET DIFFERENCIATION LYMPHOCYTAIRE T
PRECOCE
II-1. Les modèles de l’hématopoïèse
L’hématopoïèse est le processus assurant la production, le développement et la
maturation des cellules sanguines qui se déroule principalement dans la moelle osseuse. Les
travaux accumulés ces quarante dernières années ont progressivement mis en évidence une
cellule souche hématopoïétique (CSH) pluripotente proliférant et se différenciant
progressivement en précurseurs hématopoïétiques de plus en plus engagés dans une voie de
différenciation pour générer les cellules hématopoïétiques matures. Ces cellules souches
primitives sont dotées de deux propriétés principales : l’auto-renouvellement c’est-à-dire la
capacité de se multiplier et de générer une cellule fille identique à elle même à l’infini, et la
pluripotentialité qui est la capacité de générer l’ensemble des lignées hématopoïétiques
érythocytaire, mégacaryocytaire, granulocytaire et lymphocytaire. Elles ont une capacité de
reconstitution complète de l’hématopoïèse après irradiation. Chez la souris, l’isolement et la
purification des CSH repose sur l’analyse de certains marqueurs membranaires : elles
présentent le phénotype suivant, Lin-Thy-1-c-kit+Sca-1+. Chez l’homme, les CSH sont Lin-
Thy-1+CD34+CD38-. La cellule souche primitive génère les progéniteurs qui sont non
reconnaissables morphologiquement. Les progéniteurs ont une capacité d’auto-
renouvellement diminuée et un potentiel de différenciation limité à une ou deux lignées. Ils
sont désignés par leur capacité à former des colonies en milieu semi-solide : CFU (colony
forming unit) suivi des types cellulaires pouvant être générés : CFU-GEMM (granuleux,
érythrocytes, monocytes, mégacaryocytes), CFU-GM (granuleux, monocytes), BFU-E (burst
forming unit-érythrocytes), CFU-E (érythrocytes) et CFU-MK (mégacaryocytes). Puis, la
restriction du potentiel de différenciation se poursuit au niveau des précurseurs qui sont
limités à un type cellulaire et identifiables morphologiquement sur un myélogramme. La
maturation et la prolifération de ces précurseurs se poursuivent jusqu’à l’acquisition des
fonctions cellulaires spécifiques. Enfin, les cellules matures passent dans la circulation
sanguine.

28
Deux modèles d’hématopoïèse précoce sont actuellement proposés (figure 4).

29

30
Le premier modèle, défendu par l’équipe de Weissman, repose sur la séparation
précoce entre les lignées myéloïdes et lymphoïdes. Les similarités entre les lymphocytes B et
les lymphocytes T (utilisation de la même machinerie moléculaire pour les réarrangements des
immunoglobulines et du TCR, absence concomitante de ces 2 types cellulaires chez la souris
SCID) ont conduit à l’idée qu’ils dérivaient d’un progéniteur lymphoïde commun (common
lymphoid progenitor, CLP), donnant naissance aux lignées T, B et NK, indépendant du
progéniteur myéloïde commun (common myeloid progenitor, CMP) qui assure la production
des lignées granuleuse, érythrocytaire et mégacaryocytaire. L’existence du CLP a été mise en
évidence lors d’expériences de transfert chez la souris adulte [48]. Ces travaux ont montré
qu’une sous-population de cellules souches hématopoïétiques Lin-IL7R+Thy-1-Sca-1low c-
kitlow isolée de la moelle osseuse de souris adultes possédait une capacité rapide de
reconstitution des lignées lymphoïdes T, B et NK mais sans potentiel de différenciation vers la
lignée myéloïde. De la même façon, la fraction IL7R-Lin-Sca-1-c-kit+ contient des
progéniteurs à potentiel myéloïde sans potentiel lymphoïde correspondant aux CMP [49]. Ces
données suggèrent que l’engagement vers les lignées myéloïdes et lymphoïdes sont des
évènements mutuellement exclusifs.
Le second modèle, récemment développé par l’équipe de Katsura, remet en question la
ségrégation précoce entre CLP et CMP [50]. Ce modèle repose sur un nouveau système de
culture in vitro dans lequel le potentiel de différenciation de progéniteurs individuels (p) vers
les lignées myéloïdes (M), lymphocytaires T (T) et B (B) peut être analysé (multilineage
progenitor-myeloide T B, « MLP-MTB assay ») [51]. La détermination du potentiel de
différenciation cellulaire de diverses populations du foie fœtal murin par ce système permet de
détecter des progéniteurs p-MT et p-MB. En revanche, le type p-TB n’est jamais détecté dans
cet essai, indiquant que les p-T et p-B sont essentiellement produits par l’intermédiaire des
stades p-MT et p-MB respectivement. De plus, il a été montré que les p-T se développaient
plus précocément que les p-B, indiquant des voies de différenciation indépendantes [52]. Le
développent d’un autre système de culture couvrant les lignées M, érythroïde (E), T et B
(« MLP-METB assay ») a permis de montrer l’existence d’un progéniteur ayant un potentiel
myéloïde, érythroïde, lymphoïde T et B, considéré comme un progéniteur commun myéloïde
et lymphoïde (common myeloid lymphoid progenitor, CMLP) [53]. Sur la base de ces
observations, un modèle d’hématopoïèse précoce alternatif à celui communément admis
jusqu’à présent, peut être envisagé, représenté figure 4, en bas. Ce modèle est renforcé par

31
une étude récente qui a comparé le potentiel lymphoïde par culture et analyse du profil
d’expression génique par micro-array d’une nouvelle population de progéniteurs
hématopoïétiques issu du sang de cordon ombilical humain CD34+CD45RAhiCD7+ avec celui
de la population qui correspondrait aux CLP, CD34+CD45RAhiLin-CD10+ [54]. La population
CD34+CD45RAhiLin-CD10+ montre un potentiel essentiellement B, avec un profil de
transcription typique des cellules pro-B et sans expression de gènes non affiliés à la lignée B,
tandis que la population CD34+CD45RAhiCD7+ a un potentiel T et NK en culture confirmé
par le profil d’expression génique qui montre en outre l’expression de gènes des lignées
granuleuse et monocytaires. Ces données indiquent que ces 2 populations sont polarisées soit
vers les lignées T et NK, soit vers la lignée B, ce qui est en faveur d’une séparation précoce
des progéniteurs B et T/NK, ces derniers conservant un potentiel myéloïde.
II-2. Le thymus : organe de la lymphopoïèse T
Contrairement aux autres lignées cellulaires, la lignée lymphoïde T ne mature pas dans
la moelle osseuse mais dans le thymus, organe spécialisé dans la différenciation des
lymphocytes T.
Le thymus est logé dans le thorax au-dessus du cœur et des gros troncs aortiques. Il est
indispensable à la lymphopoïèse T, comme en témoigne le déficit immunitaire sévère qui
accompagne l’athymie congénitale du syndrome de Di George. Il subit une involution au
cours de l’âge. Son poids d’environ 15g à la naissance régresse proportionnellement par
rapport au poids total jusqu’à la puberté pour atteindre 20-30g. L’involution thymique
s’accélère ensuite et se poursuit tout au long de la vie. Il est cependant admis que la
production de lymphocytes T naïfs par les reliquats de tissu thymique se poursuit tout au long
de la vie mais à des niveaux de plus en plus faibles.
Le thymus est un organe lobulé dont chaque lobule comporte une région centrale, la
médullaire, entourée de cortex, lui-même subdivisé en cortex profond et superficiel. Les
précurseurs sanguins pénètrent dans le thymus au niveau de la jonction cortico-médullaire,
puis empruntent un trajet les menant du cortex superficiel vers le cortex profond, la jonction
cortico-médullaire puis la médullaire, avant leur sortie définitive au niveau des veinules de la
jonction cortico-médullaire, sous forme de lymphocytes T matures.
Le thymus est dit « lymphoépithélial » car hormis les précurseurs T, le constituant

32
principal du parenchyme thymique est la cellule épithéliale thymique, à l’origine de la plupart
des signaux de maturation que reçoit le thymocyte. Les autres populations cellulaires
présentes sont des cellules dendritiques et des macrophages chargés de l’élimination des
nombreuses cellules entrant en apoptose dans le tissu thymique.
Dans le thymus se produisent les évènements de réarrangement des gènes pour le
récepteur T à l’antigène (T cell receptor, TCR). Le réarrangement des gènes pour les
différentes chaînes des TCR αβ ou γδ génère des récepteurs à l’antigène variables d’une
cellule à l’autre, ce qui aboutit à l’élaboration à l’échelon de la population T, d’un répertoire
de lymphocytes T assez large pour reconnaître tout antigène potentiel. Se produisent ensuite
les processus de sélection positive et négative qui s’assurent que le TCR est fonctionnel, c’est-
à-dire capable de reconnaître les antigènes présentés sous forme de complexe en association
avec les molécules HLA, et qu’il n’a pas une affinité trop forte pour les peptides du soi.
II-3. Etapes précoces de la lymphopoïèse T
La différenciation lymphocytaire T thymique aboutit à la génération, la prolifération et
enfin la sélection des différentes sous populations lymphocytaires fonctionnelles. Ce
processus complexe est relativement bien étudié chez l’homme grâce aux différents
marqueurs membranaires et cytoplasmiques et aux réarrangements ordonnés et successifs des
gènes du TCR.
L’étude de nombreux marqueurs de surface, notamment grâce aux techniques de
culture thymique in vitro ou par des greffes xénogéniques chez la souris immunodéprimée
athymique (NOD-SCID), ont permis de proposer une série d’étapes de différenciation [55]
(figure 5). Elles décrivent la perte séquentielle des marqueurs d’immaturité comme le CD34
et le CD33 et le gain progressif de marqueurs T précoces : cCD3, CD7, CD2, CD5 et CD1a
puis tardifs comme le complexe CD3/TCR. Les modèles les plus récents de l’ontogénie T
humaine reconnaissent un progéniteur thymique multipotent capable de générer des
lymphocytes T, mais également des cellules NK, dendritiques et probablement monocytaires.
Ce progéniteur thymique multipotent est triple négatif CD3/4/8- et exprime le CD34, et de
manière intéressante, les marqueurs dits « myéloïdes » comme le CD13 ou le CD33. Ce
précurseur thymique multipotent n’exprime par ailleurs aucun des marqueurs T précoces
comme le CD2, le CD5 ou le CD1a. Il exprime par contre fortement le CD7, le CD45RA et le

33
CD3 cytoplasmique (cCD3). Ces caractéristiques phénotypiques et l’absence de potentiel de
différenciation érythroblastique et mégacaryocytaire le différencient nettement de la cellule
souche médullaire. Ces précurseurs CD34+, CD1a- présentent une configuration germinale de
leurs loci des TCR γ et β alors que des réarrangements incomplets du TCRδ de type Dδ2-Dδ3
ou Dδ2-Jδ1 peuvent être observés [56]. L’apparition du marqueur CD1a, qui est précédée par
l’apparition de marqueurs T comme le CD5 ou le CD2, est une étape importante dans la
maturation thymique et s’accompagne de la disparition du potentiel dendritique et dans une
moindre mesure NK. Les précurseurs CD34+, CD1a+ présentent par ailleurs des
réarrangements du TCRγ (TCRβ toujours en configuration germinale). La disparition du
CD34 s’accompagne par la suite de l’apparition du CD4 et définit la cellule CD4 immature
simple positive (CD4ISP).
Figure 5. Les différents stades de différenciation lymphocytaire T (d’après Spits, 1998) [55]
C’est au cours de cette transition que se fait l’engagement T définitif avec abandon total de
toute autre potentialité de différenciation. Cet engagement est concomitant avec le

34
réarrangement du locus β qui va aboutir à la sélection β au sein des cellules double positives
CD4+CD8+ (DP).
II-4. ββββ-sélection
Cette étape déterminante de la lymphopoïèse T αβ se produit au stade DP, CD1a+.
Elle se caractérise par l’apparition d’un Pré-TCR durant la transition CD4 ISP vers le stade
"early" DP (CD4+, CD8α+/β-). Le Pré-TCR continue à être exprimé sur les cellules DP
jusqu'à l'apparition d'un TCRαβ qui met fin au processus de β sélection.
II-4-1. Le Pré-TCR
En l’absence de la chaîne α du TCR, la chaîne β est exprimée à la surface de la cellule
DP, associée par un pont disulfure à une glycoprotéine transmembranaire de 33 Kd appelée
pré-Tα (pTα) [57, 58]. Ce complexe constitue le pré-TCR qui est associé de manière non
covalente aux chaînes ε, γ, et δ du CD3. Le pré-TCR est exprimé à la surface en très faible
quantité. Il délivre un signal de prolifération et de survie indispensable à la maturation T αβ.
Les cellules qui ne possèdent pas de réarrangement productif du TCRβ ne peuvent ainsi
former un pré-TCR et sont éliminées par apoptose. Cette étape constitue donc un "check-
point" assurant l'élimination des thymocytes engagés vers la lignée αβ mais n'ayant pas réussi
à exprimer une chaîne β fonctionnelle et à l'inverse l'amplification de celles qui ont un
réarrangement β productif.
Le pré-TCR, et donc la chaîne β du TCR, jouent donc un rôle majeur dans la
différenciation T αβ. De nombreuses données expérimentales valident cette observation. Les
souris SCID et les souris RAG-/- sont incapables d’effectuer les réarrangements des gènes des
TCR et des immunoglobulines, et n’ont pas de lymphocytes B et T matures. Le
développement lymphocytaire T est bloqué chez ces souris au stade DN. Chez les souris SCID
ou RAG-/- transgéniques pour un gène TCRβ réarrangé de façon productive, on observe
l’apparition de thymocytes DP et l’expansion du nombre de cellules dans le thymus. D’autre
part, la chaîne TCRβ est responsable de l'exclusion allélique puisque l'introduction chez la
souris d'un transgène TCRβ réarrangé de façon productive inhibe les réarrangements des loci
TCRβ endogènes [59].

35
II-4-2. Pré-Tα (α (α (α (pTα)α)α)α)
La protéine pTα est formée de plusieurs régions fortement homologues entre l’homme
et la souris: un domaine extra cellulaire de type immunoglobuline avec un pont disulfure
intra-chaîne, un peptide de connexion contenant une cystéine impliquée dans la formation du
pont disulfure avec la chaîne β, une région transmembranaire hydrophobe contenant des
résidus polaires impliqués dans l’association au CD3, et une queue intra-cytoplasmique de 31
acides-aminés chez la souris avec deux sites potentiels de phosphorylation par la Protéine
kinase C (PKC) (figure 6). Chez l’homme, cette région cytoplasmique est plus longue (114
résidus) et contient trois sites potentiels de phosphorylation par la PKC. [60].
Le gène pTα est situé sur le bras court du chromosome 6 chez l’homme (sur le
chromosome 17 chez la souris), à proximité des gènes du CMH. Il est normalement exprimé
dans le thymus de souris RAG-/- démontrant que son expression ne nécessite pas de
réarrangement. L'ARNm de pTα est exprimé avant le réarrangement β et peut donc être
détecté aux stades très précoces mais à des niveaux faibles. Son expression maximale est
atteinte au moment de la β sélection aux stades CD4ISP et DP [61]. Il n'est pas exprimé par
les thymocytes SP, ni par les cellules B, NK, myéloïdes et dendritiques [60, 61]. Cependant,
son expression est retrouvée dans les CD34+ périphériques (après mobilisation par G-CSF) et
médullaires chez l'adulte et dans les CD34 du sang de cordon et du foie fœtal [61]. De plus,
plusieurs auteurs rapportent la faible expression du pTα par des cellules cCD3+ périphériques
ou hépatiques [62, 63].
L'inactivation du gène pTα par recombinaison homologue (pTα-/-) chez la souris
aboutit à une diminution importante du nombre des thymocytes. Le blocage n'affecte que la
lignée αβ, avec un arrêt au stade DN tandis que les DP et les SP sont quasiment absents. Ceci
confirme le rôle du pré-TCR dans le passage au stade DP et dans l’expansion des DP. La
lignée γδ n'est pas affectée (voire augmentée), démontrant l'absence de rôle joué lors de cette
différenciation par le pré-TCR [64]. Il a d'ailleurs été suggéré que le pré-TCR exercerait un
rôle de contrôle négatif sur l'expression des chaînes δ et γ [57].

36
Figure 6. Représentation schématique du complexe pTαααα/TCRββββ (d’après Von Boehmer et Fehling, 1997) [65]
II-5. Réarrangement des loci du TCR
II-5-1. Définitions
Le système immunitaire se caractérise par sa capacité à reconnaître spécifiquement une
grande diversité de motifs protéiques (antigènes) et les combattre lorsqu’ils n’appartiennent
pas au soi. Cette reconnaissance se fait grâce au récepteurs présents sur les lymphocytes : le
récepteur des lymphocytes T (TCR) et le récepteur des lymphocytes B (BCR). Chaque
récepteur possédant des séquences protéiques spécifiques impliquées dans la reconnaissance
de l’antigène. Cette spécificité du récepteur définit la notion de clone pour l’ensemble des
cellules issues d’une cellule mère et exprimant le même récepteur.
Le TCR, exprimé à la surface des lymphocytes T, est un hétérodimère glycoprotéique
transmembranaire constitué de deux chaînes (αβ ou γδ) unies par un pont disulfure et
associées de façon non covalente aux chaînes CD3 γ, δ, ε, ζζ. Chaque chaîne du TCR
comporte un domaine variable (V) et un domaine constant (C), caractéristiques de la
superfamille des immunoglobulines (Ig). Les deux types de TCR αβ ou γδ sont exprimés de
P : sites potentiels de phosphorylation R : arginine/K : lysine = acides aminés polaires
Sites de glycosylation S-S : pont disulfure

37
façon mutuellement exclusive sur les lymphocytes T. La majorité des lymphocytes
périphériques (95%) exprime le TCRαβ dont le site quasi exclusif de production chez
l’homme est le thymus. Une proportion très minoritaire des populations T circulantes et
thymiques exprime le TCRγδ. Ce dernier est capable de reconnaître des antigènes non
associés au CMH. La majorité des lymphocytes TCRγδ se développe de manière thymo-
indépendante et n’exprime ni le CD4 ni le CD8. Les caractéristiques fonctionnelles des
lymphocytes T γδ sont moins bien connues mais ils semblent présenter des propriétés qui leur
sont spécifiques, liées notamment à leur localisation préférentielle au niveau des surfaces
épithéliales [66].
II-5-2. Organisation des gènes du TCR
Les gènes qui codent pour chacune des chaînes du TCR présentent une organisation
similaire et comparable à celle des gènes des Ig. Ils subissent un réarrangement somatique
avant de devenir fonctionnels. Chez l’homme comme chez la souris, les loci des gènes du
TCR ont des localisations chromosomiques différentes, excepté pour le locus δ qui est
imbriqué dans le locus α. Ainsi un réarrangement du locus α aboutit toujours à une délétion
complète du locus δ. Les gènes codant pour les TCRα/δ, β et γ sont localisés respectivement
en 14q11, 7q35 et 7p15 chez l’homme. Chaque gène comporte une partie variable en amont et
une partie constante en aval. La partie variable résulte de l’assemblage, par un processus de
recombinaison somatique médié par le complexe RAG (recombinase activating gene), des
différents segments géniques : V (Variable), D (Diversité, uniquement pour les chaînes β et
δ), et J (Jonction), séparés par de l’ADN intronique sur des distances importantes dépassant
parfois plusieurs milliers de bases. Leur recombinaison crée une séquence unique clono-
spécifique (voir III-5-3).
Nous nous intéresserons plus particulièrement aux locus δ et γ qui sont les plus
précocement réarrangés au cours de l’ontogénie T.
II-5-2-1. Locus δ
Les segments du gène du locus δ sont situés sur le chromosome 14q11 entre les
segments Vα et les segments Jα α α α (figure 7), de telle sorte q’un réarrangement Vα-Jα aboutit à

38
une délétion complète de ce locus. Il comprend un seul segment constant Cδ, 4 Jδ et 3 Dδ et 6
segments Vδ (Vδ 1, 2 et 3 étant les plus utilisés).
II-5-2-2. Locus γ
Il est localisé sur le bras court du chromosome 7 (7p15) (figure 7). Il est constitué d’un
groupe de 14 segments Vγ, situé en amont d’un groupe de 5 segments Jγ eux même organisés
en tandem avec les segments Cγ. Les segments D n’existent pas dans le locus γ. Les segments
Vγ sont groupés en 4 sous groupes : les 9 segments les plus en amont sont très homologues et
donc groupés en une même famille : VγfI. Les segments Vγ1, Vγ6 et Vγ7 sont des
pseudogènes ; seuls sont fonctionnels les segments Vγ2, Vγ3, Vγ4, Vγ5 et Vγ8. Les trois
autres familles (VγfII, III et IV) comprennent chacune un seul membre Vγ9, Vγ10 et Vγ11
respectivement. Vγ10 et Vγ11 sont également non fonctionnels du fait de l’absence de site de
splicing de leur intron leader. Les segments Jγ sont subdivisés en deux sous-groupes. En
amont du segment Cγ1, sont localisés les trois segments JP1, JP et Jγ1 ; les deux autres
segments JP2 et Jγ2 sont localisés en amont de Cγ2. Il existe une forte homologie entre JP1 et
JP2 d’une part et les segments Jγ1 et Jγ2 d’autre part. Les réarrangements impliquant les
segments proximaux, Jγ (JP et JP1/2) et Vγ (Vγ9/10/11), sont beaucoup plus fréquents parmi
les réarrangements γ durant la vie fœtale, et sont de ce fait considérés comme des
réarrangements immatures par rapport aux réarrangements impliquant les segments distaux
(Jγ1/2 et VγfI) [67].
Figure 7. Représentation schématique des loci des TCRαααα/δδδδ et γγγγ

39
II-5-3. Mécanisme de la recombinaison V(D)J et rôle de RAG
II-5-3-1. Processus de recombinaison
Le processus de recombinaison V(D)J assemble les segments discontinus variable V,
diversité D (uniquement pour TCRδ et TCRβ), et jonction J de chaque locus du TCR afin de
produire les récepteurs clono-spécifiques des lymphocytes T [68]. La recombinaison V(D)J
est dirigée par des séquences de signal de recombinaison ou RSS (Recombinaison Signal
Séquence) consistant en des motifs heptamères et nonamères conservés, séparés par des
espaces de 12 ou 23 paires de bases (pb) qui flanquent tous les segments V, D et J. La forme
consensus d’une RSS est 5’-(séquence codante)-CACAGTG-(12 ou 23 paires de bases)-
ACAAAAACC-3’. La recombinaison s’effectue généralement entre un segment génique
flanqué par un RSS avec un espace de 12 pb et un autre bordé par un RSS avec un espace de
23 pb (règle 12-23). Les RSS sont à la fois nécessaires et suffisants pour diriger la
recombinaison V(D)J, y compris pour des substrats artificiels, et constituent les sites de
clivage pour la recombinase.
Schématiquement, la réaction de recombinaison peut être subdivisée en deux phases
(figure 8). Dans une première étape, les protéines RAG-1 et 2 reconnaissent et assemblent en
un « complexe synaptique » les RSS asymétriques de deux segments de gènes, puis induisent
une cassure double brin de l'ADN. Chaque clivage conduit à la production d'une extrémité
franche au niveau des RSS et d'une structure intermédiaire en « épingle à cheveu » au niveau
de la séquence codante. Dans une seconde étape, les cassures sont identifiées, traitées et
réarrangées par la machinerie ubiquitaire de réparation de l'ADN. De manière générale, les
extrémités codantes sont rapidement assemblées en jonctions codantes. Ce processus implique
l'ouverture de la structure en épingle et des modifications favorisant la diversité des récepteurs
antigéniques par l'addition et/ou la délétion de nucléotides. En effet, des nucléotides P
(palindromiques) sont ajoutés lors du clivage asymétrique des épingles à cheveux. La diversité
N (nucléotides) résulte, quant à elle, de l'élimination puis de l'ajout de nouveaux nucléotides
par la déoxynucléotidyl terminale transférase (TdT). Ces insertions N se font de façon
aléatoire, sans matrice d'ADN, mais présentent toutefois un biais pour les résidus GC qui sont
préférentiellement intégrés. Les extrémités sont ensuite liées pour former une jonction
codante. Cette étape de réparation de l'ADN, moins bien caractérisée que la précédente, a pour
acteur central le complexe Ku/DNA-PK dont l'un des rôles aux sites de lésion serait le
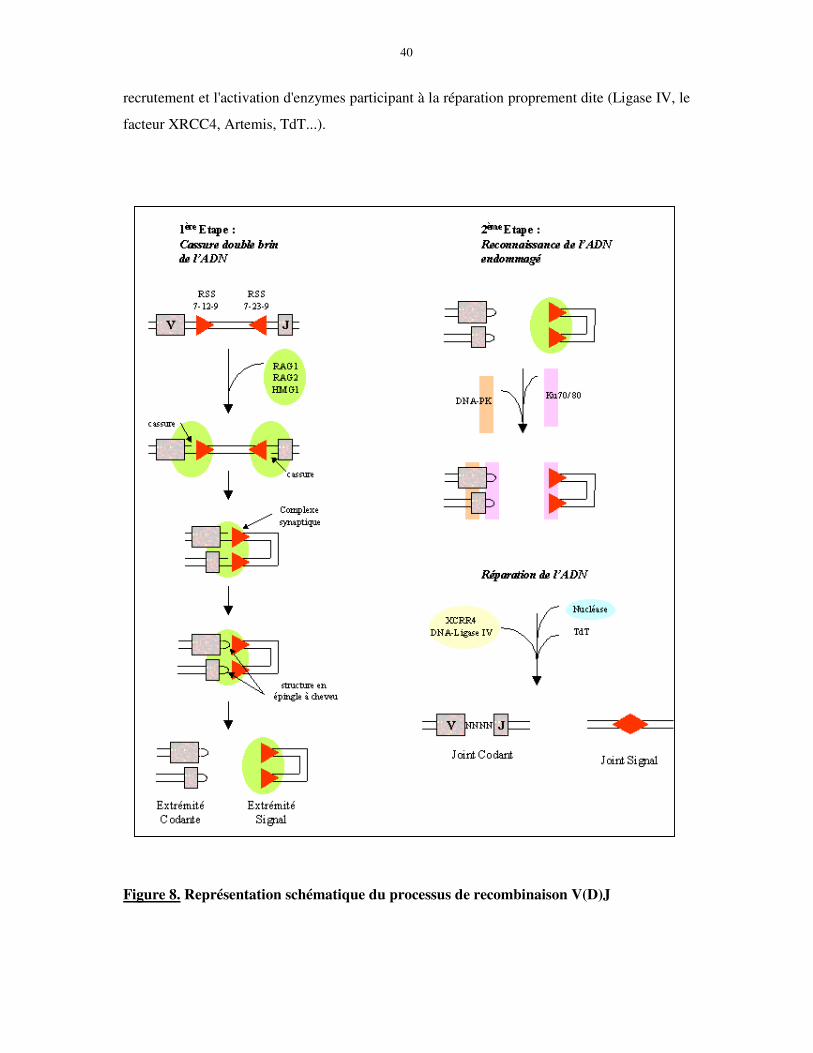
40
recrutement et l'activation d'enzymes participant à la réparation proprement dite (Ligase IV, le
facteur XRCC4, Artemis, TdT...).
Figure 8. Représentation schématique du processus de recombinaison V(D)J

41
II-5-3-2. Expression des protéines RAG1 et RAG2
Les protéines RAG1 et RAG2 sont indispensables à la recombinaison V(D)J in vivo
comme en témoigne l’absence de lymphocytes B et T matures chez les souris RAG1-/- [69].
La spécificité tissulaire de la recombinaison V(D)J est en très grande partie liée à l’expression
restreinte et très régulée des gènes RAG1 et RAG2 dans les lignées lymphoïdes B et T. Les
autres protéines impliquées dans ce processus sont des composantes du mécanisme général de
réparation de l’ADN.
Au cours de la différenciation T, les protéines RAG1 et RAG2 sont exprimées en deux
vagues successives [70]. La première vague intervient au stade DN de façon concomitante aux
réarrangements des TCR β, γ et δ. L’expression de RAG diminue ensuite lors de la β-
sélection, phase s’accompagnant d’une prolifération cellulaire. La deuxième vague coïncide
avec le réarrangement TCRα. La sélection positive met fin à l’expression de RAG.
II-5-3-3. Régulation de la recombinaison V(D)J
Les recombinaisons V(D)J sont soumises à deux types de régulation : une régulation
au cours du temps (les gènes δ se réarrangent avant les gènes γ et β, les gènes α sont les
derniers à se réarranger ; cf infra) et une régulation spécifique de lignée.
La spécificité de lignée est en partie liée à l’expression restreinte des protéines RAG1
et RAG2 aux lignées lymphoïdes. Cependant, comme la machinerie recombinase est
commune aux cellules B et T, la spécificité de lignée pourrait dépendre essentiellement de
l’accessibilité des RSS au clivage par la recombinase. Les séquences régulatrices de la
transcription, en particulier les enhancers des TCR, jouent un rôle important dans cette
accessibilité. Bien qu’il soit possible d’induire des réarrangements incomplets du TCRδ dans
des cellules humaines d’origine rénale grâce à l’expression ectopique de RAG et de facteurs
de transcription bHLH [71], les réarrangements complets et productifs ne se produisent que
dans des cellules engagées dans la différenciation T.

42
II-5-4. Cinétique de réarrangement des loci du TCR au cours de la maturation
thymique
Les réarrangements des différents loci du TCR se font selon un ordre précis lors de la
maturation thymique : δ, γ, β puis α [56] (figure 5).
Le réarrangement du locus TCRδ est le plus précoce au cours de l’ontogénie T. Parmi
les précurseurs thymique CD34+/CD1a-, 40% présentent un locus δ réarrangé tandis que les
loci γ et β sont très majoritairement en configuration germinale. Les réarrangements détectés
dans cette population sont exclusivement de type incomplets, Dδ2-Dδ3 et Dδ2-Jδ1. Il est
admis que les réarrangements du TCRδ impliquant Jδ1 nécessitent l’activation spécifique
d’enhancer T. Ainsi, les lymphocytes B peuvent réaliser des réarrangements partiels du TCRδ
de type Vδ-Dδ ou Dδ-Dδ mais pas de recombinaison impliquant les segments Jδ [72]. Ceci est
également illustré par l’absence de réarrangements du locus TCRδ impliquant Jδ1 dans les
LAL-B où les seuls réarrangements observés, dits illégitimes, du TCRδ sont de type Vδ2-Dδ3
ou Dδ2-Dδ3 [73].
Au stade CD34+/CD1a+, la grande majorité des cellules a réarrangé le locus δ.
Surviennent ensuite les réarrangements du locus TCRγ lors de la transition vers les cellules
CD4 ISP. Les segments Vγ les plus proximaux (Vγ10/Vγ11) sont les premiers utilisés.
A ces réarrangements des TCRδ et γ succèdent ceux du locus β. Ces réarrangements
sont d’abord majoritairement de type incomplet Dβ-Jβ au stade simple positif CD4 (ISP) puis
complet VDJ lors de la transition vers les cellules CD4/8 DP (EDP). Lorsqu’ils sont
productifs, ils sont suivis par l’expression de la chaîne intra cytoplasmique du TCRβ qui joue
un rôle majeur dans la différenciation αβ (voir β sélection).
Les réarrangements du TCRα surviennent plus tardivement, au stade DP. Ils se
traduisent par la délétion du locus TCRδ qui est inclus dans celui du TCRα.
II-5-5. Rôle de TEA
L’élément génique TEA (T early α) est une séquence de 380 nucléotides sans phase de
lecture ouverte localisée entre les segments Jα et le locus TCRδ (en 3’ de Cδ) [74] (figure 9)
et précédé d’un promoteur spécifique comportant des sites potentiels de fixation pour les
facteurs transcriptionnels spécifiques des lymphocytes comme TCF-1α, Ets-1 et GATA3 [75].

43
Le transcrit stérile correspondant, contenant les séquences de TEA et de Cα réunies par
épissage, est présent dans les thymocytes immatures (son expression étant maximale au stade
ISP) mais absent dans les lymphocytes matures αβ et γδ car la séquence est délétée sur les 2
allèles [76]. TEA n’est pas indispensable à la différenciation des lymphocytes αβ comme en
témoigne le développement normal de ces cellules dans les souris TEA-/-. Il a un rôle dans
l’accessibilité au locus TCRα au niveau des segments Jα les plus proximaux comme le
montre l’absence d’utilisation des segments Jα61 à Jα53 dans les lymphocytes αβ des souris
TEA-/- [77]. Le TCRα est le dernier à se réarranger et a un impact fort sur la diversification
du répertoire T final, expliquant l’absence d’exclusion allèlique, la possibilité de
réarrangements successifs et l’existence de nombreux segments Jα au sein de ce locus. TEA
permettrait l’utilisation préférentielle des segments Jα les plus proximaux afin d’éviter que le
locus soit complètement délété lors du premier réarrangement, ainsi d’autres vagues de
réarrangements sont possibles [78].
Figure 9. Localisation de la séquence TEA
IV. OBJECTIFS DU TRAVAIL
L’étude des mécanismes moléculaires de la leucémogenèse est essentielle au
développement de thérapeutiques ciblées. Un des aspects du processus de transformation
leucémique importants à comprendre est la nature de la cellule cible de la transformation
tumorale. Les progéniteurs au sein desquels surviennent les anomalies génétiques initiales
sont les « cellules souches leucémiques », à l’origine de la pérennisation du clone malin. La
détermination de leur nature est donc importante pour élaborer des approches thérapeutiques
ciblées sur elles afin de minimiser la maladie résiduelle et donc diminuer le risque de rechute.
TEA Locus TCR δδδδ Jαααα Cαααα 5 ’ 3 ’
Transcrit stérile
Vα/δα/δα/δα/δ

44
De plus, ces cellules souches leucémiques conservent en partie les caractéristiques
phénotypiques et moléculaires de la cellule normale dont elles dérivent et les transmettent à
leurs cellules filles. L’étude des cellules souches leucémiques a donc aussi un intérêt
fondamental puisqu’elle apporte des informations sur leur « contrepartie normale », et
contribue ainsi à affiner nos connaissances de l’hématopoïèse physiologique.
C’est dans cette démarche que s’inscrit notre projet dont l’objectif est d’apporter de
nouveaux éléments concernant la cible cellulaire de la translocation t(15 ;17) dans la leucémie
aiguë promyélocytaire (LAP).
La leucémie aiguë promyélocytaire est considérée comme la plus différenciée des
leucémies aiguës myéloblastiques dérivant d’un progéniteur tardif, restreint à la lignée
myéloïde. Cependant, cette hypothèse est difficilement compatible avec l’observation
qu’approximativement 25% des cas de LAP, particulièrement ceux avec la forme variante
microgranulaire, expriment le marqueur lymphocytaire T spécifique CD2. Afin de
comprendre dans quelle mesure les cellules blastiques de LAP sont apparentées à la lignée
lymphoïde T, nous avons étudié l’expression de transcrits T-spécifiques et recherché des
réarrangements des TCR dans une série de 36 cas de LAP incluant 15 formes variantes. Nous
avons choisi de quantifier pTα, RAG1 et TEA qui sont des transcrits exprimés précocement
au cours de la lymphopoïèse T et d’étudier la présence de réarrangements des TCRδ et γ qui
sont les premiers à survenir durant la maturation T. Ces données ont été comparées à celles
retrouvées dans les LAM d’autres types FAB et dans les LAL-T.

45
MATERIEL ET METHODES
I. PATIENTS
Trente-six patients porteurs d’une LAP ont été analysés ainsi que 26 cas de LAM
d’autres types FAB servant de contrôles.
Parmi les cas de LAP, 15 proviennent du service d’hématologie de l’hôpital Necker-
Enfants Malades, 15 du Guy’s Hospital de Londres et 6 du service d’hématologie du groupe
hospitalier de la Pitié-Salpétriêre. Le diagnostic cytologique fait initialement a permis de
distinguer 21 LAP de morphologie classique, hypergranulaire et 15 de forme variante
microgranulaire. Toutes les LAP étudiées présentaient la t(15 ;17)(q22 ;q21) confirmée par
méthodes cytogénétique et moléculaire. Le variant moléculaire du gène de fusion (BCR 1/2 ou
3) à été déterminé par PCR pour chaque patient. Nous disposions des données
d’immunophénotypage pour 14 des cas. L’analyse phénotypique a été faite pour 9 patients
pour lesquels nous disposions de cellules congelées. Pour tous les échantillons diagnostiques
(18 sangs et 18 moelles osseuses), la quantification des transcrits, la recherche de
réarrangement des TCR γ et δ ainsi que la détermination du statut FLT3 ont été réalisées.
Le groupe contrôle de 26 LAM d’autres types FAB comprend 5 M0, 5 M1, 4 M2, 2
M4 et 10 M5. La quantification des transcrits a été effectuée dans tous les cas (sauf pour TEA
: 25 cas/26). L’étude des réarrangements des TCR n’a pu être réalisée que chez 20 patients.
Les résultats ont été comparés à une série de 107 cas publiés de LAL-T [79].
II. PREPARATION DES CELLULES
La préparation des cellules leucémiques est réalisée à partir de cellules fraîches par
centrifugation en gradient de Ficoll, qui permet la séparation des cellules mononucléées des
hématies, plaquettes et polynucléaires. Les précurseurs granuleux appartiennent au contingent
mononucléé et sont donc récupéré dans le culot. Une fois séparées par le Ficoll, les cellules
sont lavées puis quantifiées en cellule de Malassez après coloration au bleu Trypan. Le culot

46
est conservé dans l’azote liquide en suspension volume à volume dans un milieu RPMI 1640
avec L-glutamine et 25% de DMSO.
III. IMMUNOPHENOTYPAGE
III.1. Principe de la cytométrie en flux
La cytométrie de flux est une technologie permettant la mesure simultanée des
différentes caractéristiques d’une cellule. Ces mesures sont effectuées pendant que les
cellules défilent à une vitesse de 500 à 4000 cellules par seconde dans une chambre
d’analyse dans un flux de liquide. Les cellules sont auparavant marquées par des Ac
monoclonaux liés à un fluorochrome. Le système fluidique sépare les cellules les unes des
autres, ce qui permet leur analyse une par une. La cellule passe à travers un faisceau laser
qui interagit avec celle-ci et donne des informations sur sa capacité à diffracter la lumière
incidente aux petits angles (FSC Forward Scatter : taille de la cellule) et aux grands angles
(SSC Side Scatter : complexité cytoplasmique de la cellule) et sur sa capacité à émettre une
fluorescence (si l’Ac portant le fluorochrome excité est lié à l’Ag cellulaire). Le signal
optique mesuré est converti en signal électronique proportionnel par un photomultiplicateur.
Plusieurs fluorochromes émettant à des longueurs d’onde différentes (FITC, PE,
PC5…) couplés à différents Ac sont utilisés sur une même suspension cellulaire, ce qui
permet l’analyse de la présence simultanée de plusieurs Ag cellulaires (triples voire
quadruples marquages). Le cytomètre de flux utilisé est l’appareil FACScalibur(Benton-
Dickinson).
III-2. Marquage des cellules
-Les cellules sont décongelées au bain-marie à 37°C et suspendues dans un tube contenant 2
ml de milieu RPMI. Après centrifugation 10 minutes à 1200 tours/minute, le culot est repris
dans 1 ml de milieu RPMI.
-Ensuite, les cellules sont comptées sur cellule de Malassez après coloration au bleu Trypan.
-Chaque marquage est réalisé dans des tubes Benton-Dickinson sur 400 000 cellules dans un

47
volume final de 50 µl de PBS. Plusieurs combinaisons d’anticorps marqués sont appliquées à
chaque échantillon. Les panels sont les suivants :
1/Ig2a FITC/IgG1 PE/IgG1 perCP (contrôles isotypiques)
2/CD34 FITC/CD117 PE/CD45 PerCP
3/ CD2 FITC/CD13 PE/CD45 PerCP
4/ CD5 FITC/CD19 PE/CD45 PerCP
5/ CD4 FITC/CD8 PE/CD3 PerCP
6/ HLA-DR FITC/CD33 PE/CD45 PerCP
7/ CD36 FITC/GPA PE/CD45 PerCP
8/ TdT FITC/CD79a PE/CD45 PerCP (intracytoplasmique)
9/ CD2 FITC/CD56 PE/cCD3 PerCP (intracytoplasmique)
-Pour les marquages intracytoplasmiques, une étape supplémentaire de perméabilisation est
nécessaire avant l’application des anticorps. Elle est effectuée à l’aide du kit SeraLab®.
-Après lavage, les cellules sont fixées en PFA, puis conservées à +4°C avant d’être passées au
cytomètre de flux.
III-3. Analyse de l’expression des marqueurs
Afin d’analyser la population blastique, sans tenir compte des autres types cellulaires,
il est nécessaire d’établir une « fenêtre d’acquisition » des blastes définie en fonction des FCS,
SSC et de l’expression du CD45 qui est faible comparée à celle des cellules matures. La
fluorescence est ensuite analysée à l’intérieur de cette zone d’intérêt : les résultats sont
représentés sous forme d’histogramme (intensité se fluorescence et nombre de cellules) ou de
graphe permettant de mettre en évidence des populations doublement marquées.

48
IV. QUANTIFICATION DES TRANSCRITS PAR RQ-PCR
IV-1. Extraction des ARN totaux
-Après décongélation, les cellules sont placées dans un réactif de lyse des membranes
cellulaires (RNAble, 1 ml pour 10 millions de cellules) à 4°C.
-L’extraction se fait sous hotte, dans la glace avec des tubes autoclavés et des cônes à filtre
afin d’éviter toute contamination par des ARNases risquant de dégrader l’ARN.
-Purification de l’ARN : elle est réalisée en ajoutant 100 µl de chloroforme (solvant
apolaire) par ml de lysat cellulaire. Après agitation et incubation 5 minutes dans la glace, la
phase supérieure contenant l’ARN est recueillie, sans entraîner la phase intermédiaire qui
contient l’ADN et les protéines.
-Précipitation de l’ARN : par de l’isopropanol ajouté à volume égal. Après agitation,
incubation 15 à 30 minutes à –20°C et centrifugation 15 minutes à 13 000 G, le
surnageant est éliminé et le culot d’ARN est lavé avec de l’alcool éthylique à 75%.
Après une nouvelle centrifugation suivie d’une élimination du surnageant, le culot
d’ARN séché est repris dans 15 à 30 µl d’H2O.
-La dernière étape est la mesure de la quantité d’ARN extrait. Pour cela, une mesure de la
densité optique (DO) est effectuée au spectrophotomètre à 260 et 280 nm sur l’ARN dilué au
1/400ème. La concentration est obtenue en appliquant la formule suivante : [ ] en ng/ml = DO
X 40 X400. Le rapport DO 260 / DO 280 doit être voisin de 1.8 pour que la mesure soit fiable.
-La conservation des ARN se fait à –80°C.
IV-2. Transcription Inverse
Cette technique a pour but la synthèse d’ADN complémentaire (ADNc) à partir des
ARN qui servent de matrice.
Les ARN sont décongelés sur de la glace. Un microgramme d’ARN est repris dans
6ml d’eau stérile puis incubé 10 minutes à 65°C afin de le linéariser. L’ARN est ensuite mis
en suspension dans 14µl de solution comprenant 1X de tampon, 25µM d’amorces « random
hexamere », 1µM de chaque dNTP (A, T, C, G), 20µM d’inhibiteur d’ ARNases, 10µM de
DTT (dithiothreitol, stabilisateur d’enzymes) et 100µM de transcriptase inverse du virus de la

49
leucémie murine de Moloney (M-MLV RT).
La synthèse de l’ADNc se fait selon le schéma d’incubation suivant, dans un
thermocycleur : 10 minutes à 20°C, 45minutes à 42°C puis 3 minutes à 99°C.
Les ADNc ainsi obtenus sont dilués au 2/5ème puis conservés à -20°C.
IV-3. PCR quantitative en temps réel TaqMan®
IV-3-1. Principe
Cette technique consiste en une amplification d’un fragment d’ADN entre deux
amorces spécifiques, en présence d’une sonde TaqMan® marquée par un fluorophore
« reporter » (R) et par un « extincteur » de fluorescence, le « quencher » (Q). Ce dernier
inhibe l’émission de fluorescence lorsqu’il est situé à proximité du fluorophore. Pendant la
réaction de PCR, la sonde, hybridée à l’ADN cible, est hydrolysée grâce à l’activité 5’
exonucléasique de la Taq Polymérase. La destruction de la sonde entraîne l’éloignement de R
et Q : Q ne réabsorbant plus la fluorescence, celle-ci est émise dans le milieu réactionnel
(Figure 10). L’intensité de fluorescence dans le milieu est alors proportionnelle à
l’accumulation de produits de la PCR. Une caméra mesure le spectre de fluorescence entre
500 et 650 nm, en temps réel, durant chaque élongation, sur l’appareil ABI PRISM 7000. Les
données sont intégrées par le logiciel Sequence Detector V 1.6 (Perkin Elmer/Applied
Biosystem). Il établit la fluorescence de base entre le 3ème et le 15ème cycle de PCR. La
quantification des produits de PCR se fait par la détermination d’un cycle seuil : « threshold
cycle » (Ct). Le Ct est le premier cycle pour lequel l’intensité de fluorescence est 10 fois
supérieure à la fluorescence de base. Le Ct est inversement proportionnel au nombre initial de
copies : plus ce nombre est important, plus le signal sera détecté tôt et le Ct bas.
IV-3-2. Conditions de la PCR
Le mélange TaqMan® Universal PCR Master Mix (Applied Biosystems) contient tous
les réactifs nécessaires : la Taq polymérase Gold, le tampon Gold, le MgCl2 et les
désoxynucléotides. Dans ce Master Mix, les dTTP sont remplacés par des dUTP. Cette
modification est sans effet sur l’amplification qui aboutira à la synthèse d’ADN contenant U.

50
La présence de l’ampérase Uracile N-glycosylase (UNG), active seulement en début de cycle
dans le Master Mix permet de détruire tous les brins d’ADN contenant U provenant d’une
amplification antérieure sans toucher à l’ADN de l’échantillon contenant T, évitant ainsi les
problèmes de contamination.
R : fluorophore « Reporter » Q : fluorophore « Quencher »
1. Hybridation spécifique de la sonde et des amorces sur l’ADNc 2. Elongation par la Taq polymérase et lyse de la sonde 3. Séparation de R et Q 4. Emission du signal fluorescent proportionnel à la quantité d’ADN amplifié Figure 10. Principe de la PCR quantitative par technologie TaqMan®
Le Master Mix est mélangé aux amorces spécifiques pour les gène cibles à la
concentration finale de 300 nM et à la sonde TaqMan® correspondante, à la concentration
finale de 100 nM. Cinq µl d’ADNc sont ajoutés à 20 µl de cette solution. La réaction de PCR
se fait dans des plaques de 96 puits MicroAmp optical (Perkin-Elmer/Applied Biosystems)
recouvertes d’un film plastique spécial permettant la détection de la fluorescence émise par
une fibre optique. Les produits sont amplifiés dans un ABI PRISM 7000 Sequence Detector
(Perkin-Elmer/Applied Biosystems). Les paramètres suivants sont appliqués : 1er cycle : 2
minutes à 50°C (destruction par l’UNG des éventuels produits de PCR contaminants) et 10

51
minutes à 95°C (inactivation de l’UNG et activation de la Taq polymérase), puis pour les 50
cycles suivants : 15 secondes à 95°C (dénaturation) 1 minute à 60°C (hybridation) et 1
minute à 60°C (élongation). Tous les points sont réalisés en duplicate et en intégrant au
moins 4 témoins négatifs (réactifs d’amplification sans ADNc) et 2 témoins positifs (lignée
U937 pour Abelson, lignée T HPB-ALL pour RAG1 et pTα, lignée NB4 pour TEA).
IV-3-3. Amorces et sondes
Pour chaque sonde, le quencher « extincteur » TAMRA se situe en 3’ et le marquage
fluorescent FAM en 5’ (Applied Biosystems-Perkin Elmer).
-
Transcrit Amorce sens Amorce antisens Sonde
Abelson TGGAGATAACACTCTAAGCATAA
CTAAAGGT GATGTAGTTGCTTGGGACCCA CCATTTTTGGTTTGGGCTTCACAC
CATT
pTαααα TTGGGTGTCCAGCCCTACC GCCATAGGTGAAGGCATCCA CAGCCGGCAATGGCAGTGCA
RAG1 AGCCTGCTGAGCAACCTACC GAACTGAGTCCCAAGGTGGG AGCCAGCATGGCAGCCTCTTTCC
TEA TGGAGCAGCCTGTAGCAAGA AGCTGGTACACGGCAGGGT AGTCTTGGTCCAACCAGATATCC
AGAACCCT
IV-3-4. Quantification et expression des résultats
La qualité des ARN et des cDNA est évaluée par la quantification du gène de référence
Abelson (Abl) qui est exprimé de manière ubiquitaire, constante et dont la dégradation est
similaire à celle du transcrit d’intérêt. Ainsi, les échantillons ayant des valeurs de Ct Abl
supérieures à 32 (contenant peu de transcrits d’Abl donc ayant un ARN dégradé) ont été
considérés comme ininterprétables et éliminés de la série. La moyenne des Ct obtenue pour
tous les échantillons est de 26.61.
Le résultat obtenu est corrigé grâce à l’amplification sur le même échantillon de ce
gène de référence d’expression constante : l’expression du transcrit d’intérêt est normalisé par
rapport à l’expression d’Abl afin de tenir compte de la qualité de l’ARN.
De façon très simple, le résultat de l’échantillon est représenté par un ∆Ct qui
correspond à la formule suivante : ∆Ct = Ct Abl – Ct transcrit d’intérêt. Plus le ∆Ct est élevé,
plus l’expression est importante. Une différence de 3 cycles entre 2 ∆Ct correspond à une
variation d’un log. Tous les ∆Ct inférieurs à -14 (résultats négatifs, à la limite de détection de

52
la PCR) sont arbitrairement considérés égaux à -14 afin d’éviter de tirer artificiellement les
résultats vers le bas.
V. REARRANGEMENTS DES TCR δδδδ et γγγγ
V-1. Principe
Le réarrangement des gènes du TCR dans un lymphocyte donné est unique. En effet, à
la diversité combinatoire (juxtaposition de segments V-(D)-J donnés parmi de nombreux
possibles) s’ajoute la diversité jonctionnelle (soit délétion de nucléotides, soit addition de
courtes séquences par la TdT) créant ainsi les régions « N » ou CDR3 (complementary
determining region) de séquences et de longueurs hautement variables. L’amplification de
l’ADN génomique avec des amorces spécifiques des segments V et J permet l’amplification
de cette région hypervariable. Dans une population clonale, la jonction V-(D)-J est identique
dans toutes les cellules, tandis que dans une population polyclonale, la taille des jonctions est
variable. L’analyse des produits d’amplification après électrophorèse en gel d’acrylamide, de
meilleure résolution que celle sur gel d’agarose, permet la distinction entre population clonale
(bande discrète) et polyclonale (traînée d’ADN). La qualité de l’analyse du produit de PCR
peut être améliorée grâce à l’utilisation d’amorces fluorescentes. Par cette approche, on peut
définir de manière précise la taille du réarrangement amplifié et si les amorces sont marquées
par des fluorochromes différents, les segments V, (D) ou J impliqués peuvent être identifiés.
Le contexte dans lequel nous avons appliqué cette technique est différent : le but est
d’identifier la présence d’un réarrangement dans une population blastique que nous savons
clonale et non pas de rechercher un clone dans un échantillon, mais le procédé est identique et
issu de l’action concertée du groupe BIOMED-2 BMH4-CT98-3936 [80].
V-2. Extraction d’ADN
L’extraction est réalisée à l’aide du kit Nucleon® (Amersham).
Les cellules fraîches ou décongelées à 37°C sont lavées dans 3 ml de tampon PBS
(BioMérieux). Les globules blancs sont ensuite lysés puis déprotéinisés par du perchlorate de

53
sodium. L’extraction est effectuée grâce à la résine Nucleon. Après centrifugation, la phase
supérieure contenant l’ADN est prélevée. L’ADN est ensuite précipité en ajoutant deux
volumes d’éthanol absolu froid. La méduse d’ADN est récupérée à la pipette, lavée à l’éthanol
à 70%, séchée puis dissoute dans du TE (Tris HCl pH73.6, EDTA pH8 1mM).
La concentration d’ADN est déterminée après dilution au 1/200e dans du TE, par la
mesure de la DO à 260 nm par spectrophotométrie aux ultraviolets. La pureté de l’ADN est
affirmée par un rapport (DO à 260 nm – DO à 320 nm/ DO à 280 nm – DO à 320 nm) entre
1.8 et 2.
L’ADN est conservé à 4°C.
V-3. Réarrangements du TCR δδδδ
Dans un premier temps, une PCR multiplexe est réalisée. Si elle révèle la présence
d’un réarrangement, l’identification des segments impliqués se fait dans un deuxième temps
par une PCR triplex utilisant des amorces fluorescentes.
V-3-1. PCR multiplexe
V-3-1-1. Conditions de PCR et séquence des amorces
La PCR multiplexe est réalisée en utilisant 7 amorces sens (6 reconnaissant chacune
un segment V de Vδ1 à Vδ6 et une se liant au segment Dδ2) et 5 amorces antisens
reconnaissant chacune un gène Jδ (Jδ1 à 4) et Dδ3. La localisation des amorces est représentée
sur la figure 11.
Les séquences des oligonucléotides sont celles référencées par le groupe BIOMED-2 BMH4-
CT98-3936 [80].
Cent nanogramme d’ADN est suspendu dans une solution comprenant du tampon 1X,
du MgCl2 (25mM), des dNTP (2 mM), la Taq Gold 1 U et chacune des 12 amorces à 0.2 µM
dans un volume final de 50 µl.
Le programme d’amplification est le suivant : dénaturation initiale et activation de la Taq
Gold : 94° 8 minutes, puis pour 35 cycles : dénaturation 94°C 45 secondes, hybridation 60°C
1 minute, élongation 72°C 1 minute 30.

54
PCR Multiplex
* * *
*patients positifs
PCR Triplex
Figure 11. Recherche de réarrangements du TCRδδδδ : localisation des amorces et exemple de résultat
�δδδδ���δδδδ��������� ����� ��������������
�δδδδ���δδδδ��������� ����� ��������������
Vδ δ δ δ
4
Vδ δ δ δ
6
5’
Vδ δ δ δ
1
Vδ δ δ δ
5
Vδ δ δ δ
7
Vδ δ δ δ
8
δ δ δ δ R
EC
Vδ δ δ δ
2
ϕ J
αα αα
61 J αααα (71 Kb)
Dδ δ δ δ
1D
δ δ δ δ 2
Dδ δ δ δ
3
1234
J δδδδ
Vδ δ δ δ
3C
δδ δδ
Cαα αα
20 V α 330 Kb
11 V α 160 Kb
6 Vα 60 Kb
7 Vα 65 Kb
3 Vα 55 Kb
3 Vα 135 Kb
50 Kb
←←←←
1000 Kb
D�δ2�→→→→�D�δ3�←←←←�
J�δ�1�J�δ�2�J�δ�3�J�δ�4�←←←←�V�δ1�V�δ2�
V�δ3�V�δ4�V�δ5�V�δ6�
→→→→�

55
V-3-1-2. Analyse des produits d’amplification
Les produits d’amplification sont soumis à un cycle de dénaturation-réhybridation
(100°C 3 minutes puis 65 °C 3 minutes) afin qu’il y ait formation des éventuels
hétéroduplexes.
Une électrophorèse en gel de polyacrylamide à 8% est ensuite réalisée. La révélation se
fait par coloration au bromure d’éthidium illuminé aux ultraviolets.
Plusieurs ADN témoins sont amplifiés simultanément permettant de contrôler la qualité de la
réaction. Le contrôle négatif est l’ADN issu de lymphocytes périphériques normaux
polyclonaux. Le contrôle positif est l’ADN d’une lignée lymphocytaire T (PEER) présentant
des réarrangements des TCR δ et γ de taille connue.
La présence d’une bande discrète évoque la présence d’un réarrangement qui devra
être confirmé et identifié par une deuxième amplification utilisant 3 amorces fluorescentes.
V-3-2. PCR triplex
V-3-2-1. Conditions de PCR et séquence des amorces
Trois amorces sens marquées par 3 fluorochromes différents sont utilisées (Vδ1, Vδ2
et Dδ2) ainsi que 2 amorces antisens (Dδ3 et Jδ1) reconnaissant les segments les plus souvent
impliqués dans les réarrangements du TCRδ. Les conditions de PCR sont identiques à celles
de la PCR multiplexe. Les témoins négatifs et positifs sont les mêmes que précédemment.
Les séquences des oligonucléotides sont les mêmes que précédemment, seule le
marquage par un fluorochrome diffère.
V-3-2-2. Analyse des produits d’amplification
Les produits d’amplification sont analysés dans un séquenceur (ABI prism 310) qui
permet une électrophorèse en capillaire. L’analyse de l’électrophorégramme à l’aide du
logiciel ABI Prism renseigne précisément sur la taille des fragments ainsi que sur les segments
V utilisés en fonction de la « couleur » des pics correspondant aux différents fluororochromes.
En présence d’un réarrangement monoclonal, un pic unique sera obtenu : en fonction de sa
couleur, on déterminera lequel des gènes Vδ1, Vδ2 et Dδ2 est utilisé, et en fonction de sa

56
taille, on déduira si Jδ1 ou Dδ3 est impliqué. (figure 11). L’utilisation de ces segments est
ensuite confirmée par une PCR simplex, utilisant un seul couple d’amorce correspondant aux
gènes suspectés et révélée par électrophorèse en gel de polyacrylamide.
V-4. Réarrangements du TCRγγγγ
La recherche de réarrangements du TCRγ se fait par 2 PCR multiplexes utilisant des
amorces fluorescentes.
V-4.1. Conditions de PCR et séquence des amorces
Deux mélanges d‘amorces sont utilisés (1 et 2). Ils ont en commun les 3 amorces
fluorescentes antisens reconnaissant Jγ1/2, JP1/2 et JP (figure 12). Le premier mélange
contient une amorce consensus reconnaissant les gènes Vγ de la famille I (VγfI) et une
amorce reconnaissant Vγ10. Le deuxième mélange contient 2 amorces reconnaissant V9 et
V11.
Les séquences des oligonucléotides sont celles référencées par le groupe BIOMED-2 BMH4-
CT98-3936 [80].
Chaque tube de PCR contient, dans un volume final de 50 µl, 1 µg d’ADN, 1X de
Tampon II (Perkin Elmer), 2.5 mM de MgCl2, 0.2 mM de dNTP, 1 U de Taq A (Perkin
Elmer) et les amorces (0.25 µM chacune sauf VγfI Vγ10 Vγ9 et Vγ11 à 0.5 µM).
Le programme d’amplification est identique pour les 2 PCR : étape initiale de 3 minutes à
92°C, 35 cycles comprenant successivement 30 secondes à 92°C, 1 minute à 57°C, 1 minute à
72°C, puis élongation finale 10 minutes à 72°C.
V-4-2. Analyse des produits d’amplification
Comme pour la PCR triplex TCRδ, les produits d’amplification sont analysés dans un
séquenceur (ABI prism 310). En fonction de la couleur du pic, on déduit lequel des segments
Jγ est utilisé puis en fonction de la taille, on détermine lequel des gènes Vγ est impliqué
(figure 12).
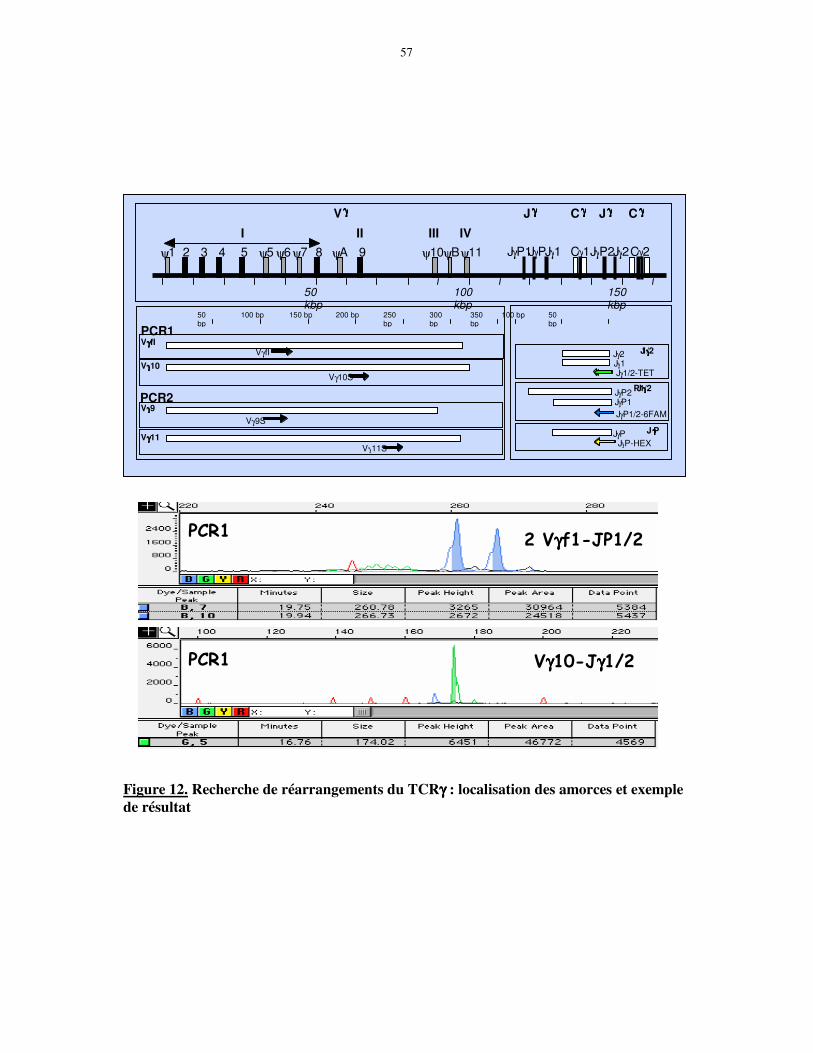
57
Figure 12. Recherche de réarrangements du TCRγγγγ : localisation des amorces et exemple de résultat
50 bp� 100 bp� 150 bp� 200 bp� 250
bp� 300 bp� 350
bp� 50 bp�100 bp�
PCR1�
ψ�1�2� 3� 4� 5� ψ�5�ψ�6�ψ�7�8�ψ�A� 9� ψ�10�ψ�B�ψ�11� J�γ�P1�J�γ�P�J�γ�1�C�γ�1�J�γ�P2�J�γ�2�C�γ�2�I� II� III� IV�
V�γγγγ� J�γγγγ� J�γγγγ�C�γγγγ� C�γγγγ�
50 kbp� 100
kbp� 150 kbp�
V�γ�fI�
V�γ�11S�V�γγγγ11�
V�γ�10S�V�γγγγ10�
V�γ�9S�V�γγγγ9�
V�γγγγfI�J�γγγγ�1/2�
J�γ�1�J�γ�2�J�γ�1/2-TET�
J�γγγγ�P1/2�J�γ�P1�J�γ�P2�J�γ�P1/2-6FAM�
J�γγγγ�P�J�γ�P�J�γ�P-HEX�
PCR2�
�����γγγγ���� �
�γγγγ���γγγγ ����

58
VI. RECHERCHE D’UNE DUPLICATION EN TANDEM DE FLT3
(FLT3-ITD)
La recherche d’une duplication des exons 11 et 12 du gène codant pour FLT3 se fait
par amplification de l’ADN complémentaire à l’aide d’amorces marquées par un
fluorochrome, suivie par une analyse en électrophorèse capillaire (GenScan) [81].
VI-1. Conditions de la PCR
Chaque tube de PCR contient, dans un volume final de 50 µl, 8 µl d’ADNc, 1X de
Tampon Gold I (Perkin Elmer), 5 mM de MgCl2, 0.75 mM de dNTP, 1.25 U de Taq Gold
(Perkin Elmer) et 0.3 µM de chacune des 2 amorces.
Le programme d’amplification est le suivant : dénaturation initiale et activation de la Taq
Gold : 94° 8 minutes, puis pour 35 cycles : dénaturation 94°C 30 secondes, hybridation 60°C
1 minute, élongation 72°C 1 minute 30 suivi d’une élongation finale de 10 minutes à 72°C. .
VI-2. Séquences des oligonucléotides
Amorce sens, FLT3E (S) TGGTGTTTGTCTCCTCTTCATTGT
Amorce antisens fluorescente: FLT3Q (AS) GTTGCGTTCATCACTTTTCCAA
VI-3. Analyse des produits d’amplification
L’analyse des produits d’amplification est réalisée par électrophorèse capillaire dans
un séquenceur automatique (ABI 310) qui permet la détermination précise de la taille des
fragments (figure 13). L’allèle sauvage, non dupliqué a une taille attendue de 238 pb. S’il
existe un allèle muté présentant une duplication, un deuxième pic apparaît de taille variable en
fonction du nombre de nucléotides additionnels (de 18 à 190 pb de plus que l’allèle normal).

59
Témoin négatif
Témoin positif Patient
Figure 13. Recherche d’une duplication de FLT3 : analyse des produits de PCR par
électrophorèse en capillaire (GenScan) sur ABI 310
En haut : témoin négatif (lignée U937), on observe un pic correspondant à la bande germinale de 238 pb Au milieu : témoin positif (lignée MV), on observe un pic de 269 pb correspondant à une duplication du gène FLT3 présente à l’état homozygote. En bas : patient, on observe 2 pics, un de 238 pb correspondant à la bande germinale, et un pic à 302 pb correspondant à une duplication du gène FLT3 présente à l’état hétérozygote

60
VII. STATISTIQUES
La comparaison entre deux pourcentages est réalisée grâce à un test de Chi2.
La comparaison des moyennes de ∆Ct est effectuée par un test de Student bilatéral.
Les différences pour lesquelles la valeur de p est inférieure ou égale à 0.05 sont considérées
comme statistiquement significatives.
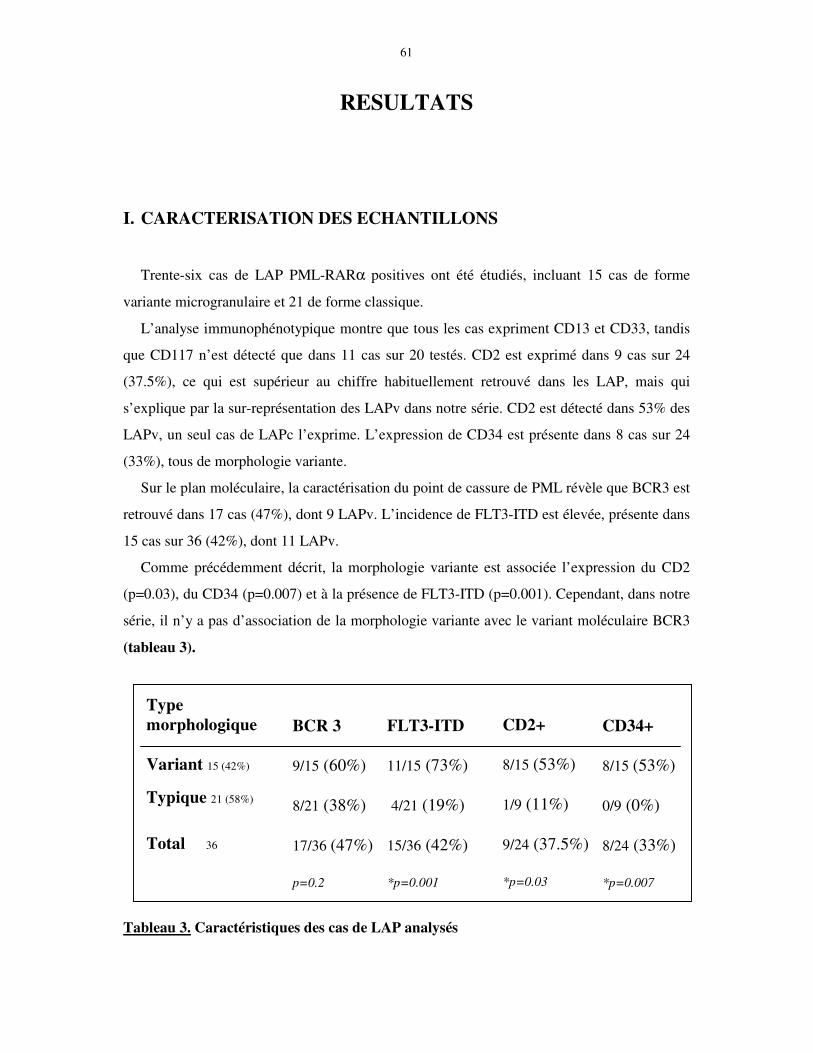
61
RESULTATS
I. CARACTERISATION DES ECHANTILLONS
Trente-six cas de LAP PML-RARα positives ont été étudiés, incluant 15 cas de forme
variante microgranulaire et 21 de forme classique.
L’analyse immunophénotypique montre que tous les cas expriment CD13 et CD33, tandis
que CD117 n’est détecté que dans 11 cas sur 20 testés. CD2 est exprimé dans 9 cas sur 24
(37.5%), ce qui est supérieur au chiffre habituellement retrouvé dans les LAP, mais qui
s’explique par la sur-représentation des LAPv dans notre série. CD2 est détecté dans 53% des
LAPv, un seul cas de LAPc l’exprime. L’expression de CD34 est présente dans 8 cas sur 24
(33%), tous de morphologie variante.
Sur le plan moléculaire, la caractérisation du point de cassure de PML révèle que BCR3 est
retrouvé dans 17 cas (47%), dont 9 LAPv. L’incidence de FLT3-ITD est élevée, présente dans
15 cas sur 36 (42%), dont 11 LAPv.
Comme précédemment décrit, la morphologie variante est associée l’expression du CD2
(p=0.03), du CD34 (p=0.007) et à la présence de FLT3-ITD (p=0.001). Cependant, dans notre
série, il n’y a pas d’association de la morphologie variante avec le variant moléculaire BCR3
(tableau 3).
Tableau 3. Caractéristiques des cas de LAP analysés
Type morphologique Variant 15 (42%)
Typique 21 (58%)
Total 36
BCR 3 9/15 (60%) 8/21 (38%) 17/36 (47%) p=0.2
FLT3-ITD 11/15 (73%) 4/21 (19%) 15/36 (42%) *p=0.001
CD2+ 8/15 (53%) 1/9 (11%) 9/24 (37.5%) *p=0.03
CD34+ 8/15 (53%) 0/9 (0%) 8/24 (33%) *p=0.007

62
II. EXPRESSION DE TRANSCRITS LYMPHOÏDES T PRECOCES
DANS LA LAP
II-1. Les blastes de LAP expriment pTαααα mais pas RAG1
La quantification des transcrits de pTα et RAG1 a été réalisée sur tous les échantillons
de LAP et dans 26 cas d’autres types de LAM. Les résultats sont exprimés en ∆Ct (Ct Abl - Ct
pTα ou RAG1) et comparés aux niveaux observés dans les 107 cas de LAL-T précédemment
publiés [79]. Ils sont considérés comme positifs si le ∆Ct est supérieur à –6, ce qui correspond
1% du niveau de transcrits observé dans la lignée cellulaire de référence HPB-ALL.
II-1-1. pTαααα
La moyenne des niveaux d’expression de pTα (figure 14) est significativement plus
élevée dans l’ensemble des LAP comparé aux autres types de LAM (-3.70 versus –11.10,
p<0.001). Les niveaux les plus élevés sont observés dans les LAPv (-2.38 contre -4.64 dans
les LAPc, p=0.049), et sont comparables à ceux retrouvés dans les LAL-T (-2.1).
Les niveaux d’expression de pTα dans les LAM contrôles sont hétérogènes : ils sont
plus élevés dans les LAM 0, 1 et 2, myéloïdes, que dans LAM 4 et 5, monocytaires (-5.7 et –
12.23, p<0.001). Nous avons donc divisé le groupe contrôle en 14 LAM myéloïdes (LAM
0/1/2) et 12 LAM monocytaires (LAM 4/5).
Le niveau moyen de pTα des LAPv est significativement plus élevé que celui des
LAM 0/1/2 (p=0.004) et des LAM 4/5 (p<0.001), tandis que les niveaux observés dans les
LAPc sont similaires à ceux des LAM 0/1/2 (p=0.31) mais supérieurs à ceux des LAM 4/5
(p<0.001).
En définissant le seuil de positivité par un ∆Ct supérieur à –6, 93% des LAPv, 76%
des LAP classiques sont pTα positives contre 50% des LAM 0/1/2 et 8% des LAM4/5
(tableau 4).

63
Figure 14. Expression de pTαααα dans les différents types de leucémies aiguës Chaque trait (-) représente la médiane avec les écart-types
II-1-2. RAG1
RAG1 n’est pas exprimé dans les LAPv ou les formes classiques, ni dans les autres
types de LAM (figure 15) contrairement aux LAL-T dans lesquelles il y a une expression
coordonnée et à des niveaux similaires de pTα et RAG1.
Au total ces données montrent que pTα est exprimée à des niveaux élevés et similaires
à ceux des LAL-T dans les LAPv, à des niveaux plus faibles mais significatifs dans les LAPc
et les LAM 0/1/2, mais pas dans les LAM 4/5. Contrairement aux LAL-T, dans les LAPv,
pTα est exprimée en l’absence de RAG1.
-16
-14
-12
-10
-8
-6
-4
-2
0
2
4
∆∆ ∆∆C
T pt
αα αα
�� ��� �� �� �� �� �� �
� �
�� � � �
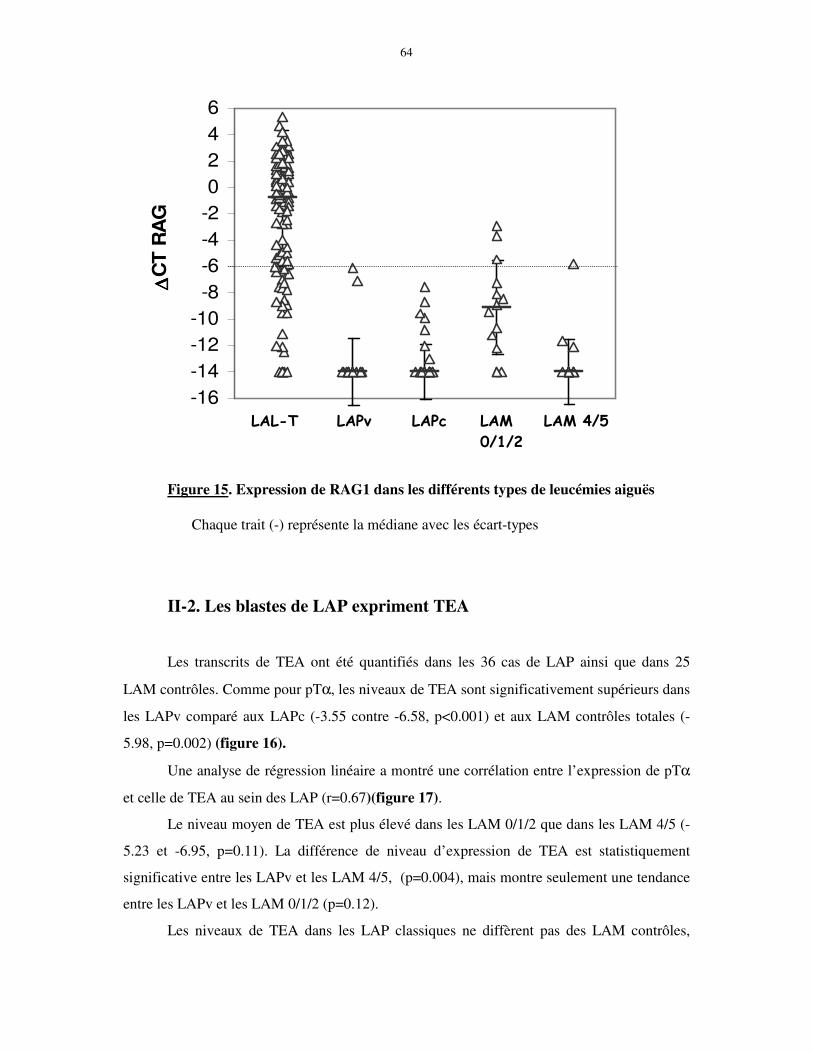
64
Figure 15. Expression de RAG1 dans les différents types de leucémies aiguës Chaque trait (-) représente la médiane avec les écart-types
II-2. Les blastes de LAP expriment TEA
Les transcrits de TEA ont été quantifiés dans les 36 cas de LAP ainsi que dans 25
LAM contrôles. Comme pour pTα, les niveaux de TEA sont significativement supérieurs dans
les LAPv comparé aux LAPc (-3.55 contre -6.58, p<0.001) et aux LAM contrôles totales (-
5.98, p=0.002) (figure 16).
Une analyse de régression linéaire a montré une corrélation entre l’expression de pTα
et celle de TEA au sein des LAP (r=0.67)(figure 17).
Le niveau moyen de TEA est plus élevé dans les LAM 0/1/2 que dans les LAM 4/5 (-
5.23 et -6.95, p=0.11). La différence de niveau d’expression de TEA est statistiquement
significative entre les LAPv et les LAM 4/5, (p=0.004), mais montre seulement une tendance
entre les LAPv et les LAM 0/1/2 (p=0.12).
Les niveaux de TEA dans les LAP classiques ne diffèrent pas des LAM contrôles,
-16-14-12-10-8-6-4-20246
∆∆ ∆∆C
T R
AG
�� ��� �� �� �� �� �� �
� �
�� � � �
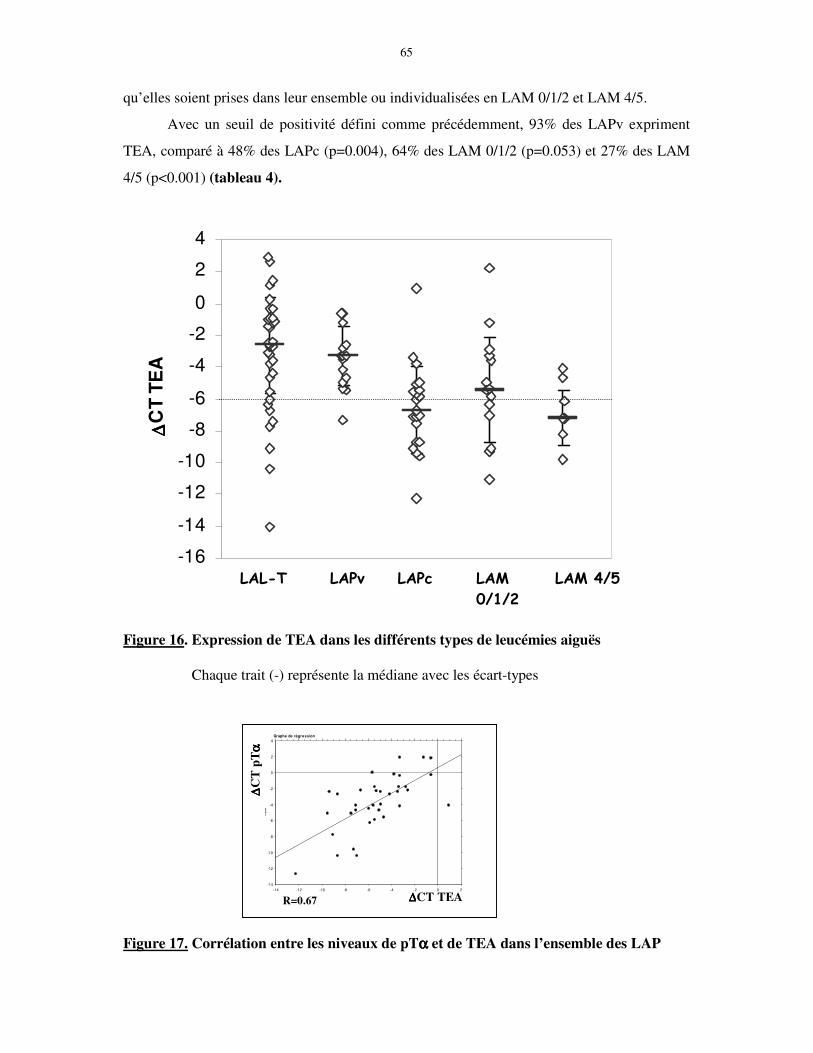
65
qu’elles soient prises dans leur ensemble ou individualisées en LAM 0/1/2 et LAM 4/5.
Avec un seuil de positivité défini comme précédemment, 93% des LAPv expriment
TEA, comparé à 48% des LAPc (p=0.004), 64% des LAM 0/1/2 (p=0.053) et 27% des LAM
4/5 (p<0.001) (tableau 4).
Figure 16. Expression de TEA dans les différents types de leucémies aiguës Chaque trait (-) représente la médiane avec les écart-types
Figure 17. Corrélation entre les niveaux de pTαααα et de TEA dans l’ensemble des LAP
-16
-14
-12
-10
-8
-6
-4
-2
0
2
4
∆∆ ∆∆C
T TE
A
�� ��� �� �� �� �� �� �
� �
�� � � �
-14
-12
-10
-8
-6
-4
-2
0
2
4
Dpt
a
-14 -12 -10 -8 -6 -4 -2 0 2Dtea
Graphe de régression
∆∆∆∆CT TEA
∆∆ ∆∆C
T p
Tαα αα
R=0.67

66
II-3. Expression de TdT et CD3 cytoplasmique
L’expression de CD3 cytoplasmique (cCD3) est retrouvée dans 2 cas sur 15 cas de
LAP analysés. Ces 2 cas présentaient une morphologie variante, un point de cassure PML
BCR3, une duplication de FLT3 et une expression relativement élevée de pTα et de TEA
(pTα : -2.3 et -2.4, TEA : -5.35 et -3.50).
L’expression de TdT intracytoplasmique est observée dans 3 cas sur 18 analysés, sans
co-expression de cCD3. Tous trois présentaient une forme variante, 2 avaient un point de
cassure PML BCR3 et une duplication de FLT3. De forts taux de pTα et TEA sont retrouvés
dans ces 3 cas (pTα : 1.9, 1.8, -1.8, TEA : -0.6, -1.25 et -2.8).
TdT et cCD3 sont donc exprimés préférentiellement dans les LAP variantes pTα+ et
TEA+.
Il est intéressant de noter que les 4 LAM0 et l’unique LAM2 testées expriment TdT et que 4/9
expriment cCD3 (1/4 M0, 2/3 M1, 1/2 M2), tandis que ces 2 marqueurs ne sont retrouvés dans
aucune des LAM 4/5.
LAP classique LAP variante
p (entre LAPc et LAPv)
p (entre LAPv et LAM 0/1/2)
LAM 0/1/2 LAM 4/5 p (entre LAM 0/1/2 et LAM4/5)
Total 21 15 14 12 BCR3 8 (38%) 9 (60%) 0.19 FLT3-ITD 4 (19%) 11 (73%) 0.001 0.01 3/12 (25%) 2/12 (17%) CD2+ 1/9 (11%) 8/15 (53%) 0.03 2/11 (18%) 1/7 CD34+ 0/9 (0%) 8/15 (53%) 0.007 6/11 (55%) 2/7 cCD3+ 0/3 (0%) 2/12 (17%) 4/9 0/7 cTdT+ 0/5 (0%) 3/13 (23%) 5/7 0/7 CD19+ 0/1 (0%) 2/13 (15%) 0/4 0/7 pTα+ 16 (76%) 14 (93%) 0.17 7/14 (50%) 1/12 (8%) 0.02 moyenne pTα�� (∆CT) �
-4.64 -2.38 0.049 0.009 -5.7 -12.23 <0.001
TEA+ 10 (48%) 14 (93%) 0.004 0.004 9/14 (64%) 3/11 (27%) 0.06 moyenne TEA (∆CT �)
-6.58 -3.55 <0.001 0.053 -5.23 -6.95 0.11
Réarrangement des TCR 1/17 (6%) 4/15 (27%) 0.11 0.12 0/13 (0%) 1/7 (14%) 0.044
Tableau 4. Tableau récapitulatif des caractéristiques des sous-groupes de LAM étudiés
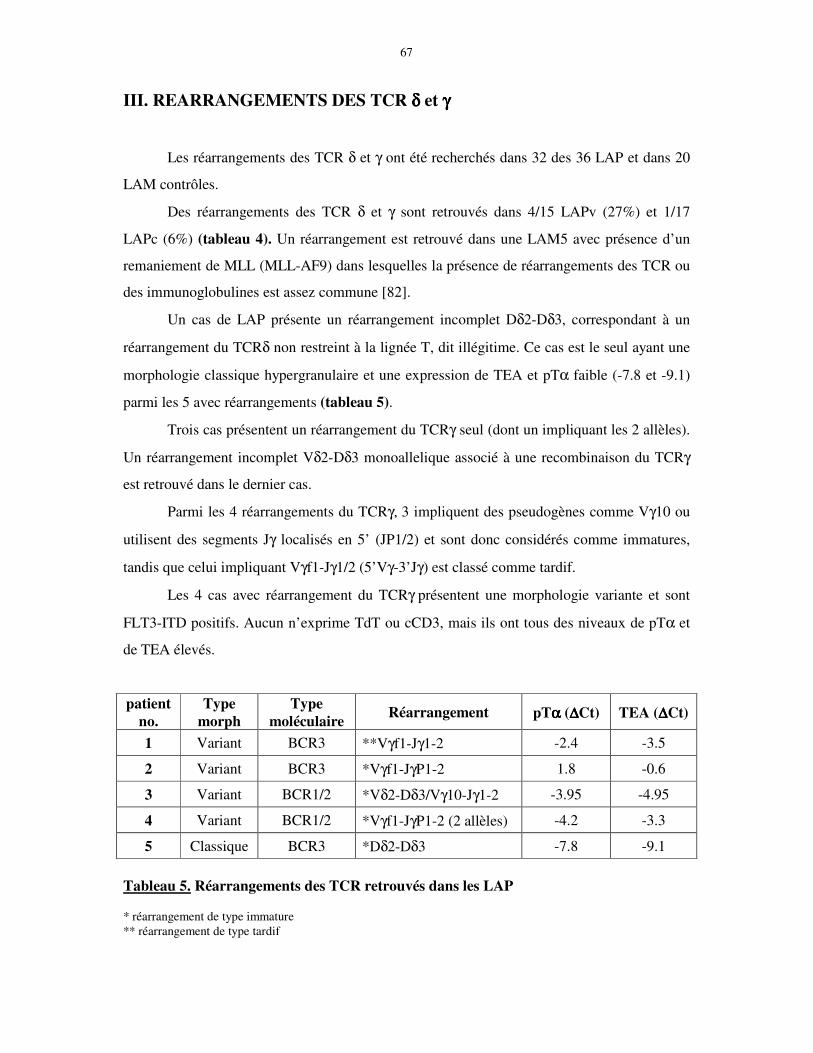
67
III. REARRANGEMENTS DES TCR δδδδ et γγγγ
Les réarrangements des TCR δ et γ ont été recherchés dans 32 des 36 LAP et dans 20
LAM contrôles.
Des réarrangements des TCR δ et γ sont retrouvés dans 4/15 LAPv (27%) et 1/17
LAPc (6%) (tableau 4). Un réarrangement est retrouvé dans une LAM5 avec présence d’un
remaniement de MLL (MLL-AF9) dans lesquelles la présence de réarrangements des TCR ou
des immunoglobulines est assez commune [82].
Un cas de LAP présente un réarrangement incomplet Dδ2-Dδ3, correspondant à un
réarrangement du TCRδ non restreint à la lignée T, dit illégitime. Ce cas est le seul ayant une
morphologie classique hypergranulaire et une expression de TEA et pTα faible (-7.8 et -9.1)
parmi les 5 avec réarrangements (tableau 5).
Trois cas présentent un réarrangement du TCRγ seul (dont un impliquant les 2 allèles).
Un réarrangement incomplet Vδ2-Dδ3 monoallelique associé à une recombinaison du TCRγ
est retrouvé dans le dernier cas.
Parmi les 4 réarrangements du TCRγ, 3 impliquent des pseudogènes comme Vγ10 ou
utilisent des segments Jγ localisés en 5’ (JP1/2) et sont donc considérés comme immatures,
tandis que celui impliquant Vγf1-Jγ1/2 (5’Vγ-3’Jγ) est classé comme tardif.
Les 4 cas avec réarrangement du TCRγ présentent une morphologie variante et sont
FLT3-ITD positifs. Aucun n’exprime TdT ou cCD3, mais ils ont tous des niveaux de pTα et
de TEA élevés.
patient no.
Type morph
Type moléculaire Réarrangement pTα α α α (∆∆∆∆Ct) TEA (∆∆∆∆Ct)
1 Variant BCR3 **Vγf1-Jγ1-2 -2.4 -3.5
2 Variant BCR3 *Vγf1-JγP1-2 1.8 -0.6
3 Variant BCR1/2 *Vδ2-Dδ3/Vγ10-Jγ1-2 -3.95 -4.95
4 Variant BCR1/2 *Vγf1-JγP1-2 (2 allèles) -4.2 -3.3
5 Classique BCR3 *Dδ2-Dδ3 -7.8 -9.1 Tableau 5. Réarrangements des TCR retrouvés dans les LAP * réarrangement de type immature ** réarrangement de type tardif

68
IV. EXPRESSION DE pTα α α α ET TEA EN FONCTION DES SOUS-
GROUPES de LAP
Afin de déterminer si l’expression de pTα et de TEA est corrélée à des paramètres
autres que la morphologie variante, nous avons comparé les niveaux de transcrits au sein des
36 LAP classées en fonction de leur morphologie, de la présence de FLT3-ITD et de
l’expression de CD2, CD34 ou CD117 (tableau 6). L’analyse statistique (test de Student)
révèle que l’expression de pTα est corrélée à celle du CD34 (p=0.03).
TEA n’est corrélée de manière significative qu’avec la morphologie variante (p<0.001).
Il est intéressant de noter que ni pTα, ni TEA n’est corrélé avec l’expression de CD2.
pTαααα TEA
Sous-groupe + - p + - p
LAPv -2.38 -4.64 0.049 -3.55 -6.58 <0.001
BCR3 -3.57 -3.81 0.83 -5.38 -5.26 0.89
FLT3-ITD -3.65 -3.73 0.94 -4.83 -5.75 0.33
CD2 -3.61 -3.73 0.93 -4.88 -4.49 0.75
CD34 -1.73 -4.86 0.03 -3.27 -5.32 0.10
CD117 -2.29 -4.21 0.18 -3.71 -3.75 0.97
Tableau 6. Moyennes des ∆∆∆∆Ct de pTαααα et TEA dans les différents sous-groupes de LAP

69
DISCUSSION
L’ensemble de nos résultats montre que les formes variantes de LAP expriment des
transcrits pTα et TEA, en association à d’autres marqueurs d’orientation précoce vers la
lignée lymphoïde T, comme l’expression de CD2, cCD3 ou TdT et la présence de
réarrangements du TCR. Ces données s’intègrent difficilement dans l’hypothèse de la
survenue de la t(15 ;17) dans un progéniteur engagé dans la lignée myéloïde et suggèrent, soit
que ces transcrits aberrants sont le résultat d’une dérégulation directe ou indirecte médiée par
PML-RARα (infidélité de lignée), soit que la cellule souche leucémique dans la LAP variante
a un potentiel à la fois myéloïde et lymphoïde T (promiscuité de lignée).
I. INFIDELITE DE LIGNEE
Le processus leucémique peut s’accompagner de la dérégulation de l’expression des
gènes et entraîner ainsi l’expression aberrante de marqueurs présents habituellement dans
d’autres lignées que celle dont dérive la population blastique. C’est la théorie de l’infidélité de
lignée [83]. Il est possible que l’acquisition des caractéristiques de la lignée lymphoïde T dans
les LAPv résulte de la dérégulation de l’expression des gènes par la protéine de fusion PML-
RARα.
PML-RARα peut avoir une action directe sur l’expression des gènes affiliés à la lignée
T. La dérégulation de l’expression des gènes peut aussi être secondaire à une instabilité
génomique liée à la présence de PML-RARα..
I-1. Action directe de PML-RARαααα sur l’expression des gènes affiliés à
la lignée T
Il est possible que l’expression des gènes affiliés à la lignée T dans les LAPv résulte de
la liaison directe de la fusion PML-RARα sur une séquence régulatrice des gènes pTα et
TEA, bien qu’elle soit plutôt un répresseur transcriptionnel.

70
Cependant, nous n’avons pas identifié de motif de fixation aux récepteurs de l’acide
rétinoïque (séquence RARE) dans les régions conservées du promoteur et des exons 1 et 2 du
gène pTα.
De plus, aucune donnée de la littérature ne permet d’impliquer la voie de l’AR dans la
différenciation lymphocytaire T. La perte d’expression de RARα/RXR n’a pas d’influence sur
le développement T [20].
Enfin, la protéine de fusion PML-RARα étant présente dans les 2 formes de LAP (classique et
variante) sans différence de points de cassure au niveau de PML dans notre série, et sans
corrélation entre le point de cassure et l’expression de pTα/TEA, l’effet direct de PML-RARα
sur l’expression des gènes T ne semble pas pouvoir expliquer la différence entre les deux
formes.
I-2. Instabilité génomique et dérégulation secondaire de l’expression
des gènes
La dérégulation de l’expression des gènes pourrait survenir dans un contexte d’instabilité
génomique induisant des évènements secondaires, incluant une dérégulation de la chromatine
aboutissant à l’expression aberrante de protéines.
I-2-1. Nécessité d’évènements secondaires pour aboutir au phénotype leucémique
complet
Bien que la fusion PML-RARα soit nécessaire, elle n’est pas suffisante au
développement d’une LAP chez les souris transgéniques. En effet, dans ces modèles murins,
la LAP survient après une longue période de latence et avec une faible fréquence [29-31]. La
nécessité d’évènements oncogéniques additionnels est démontrée par une pénétrance accrue
du phénotype leucémique et une période de latence raccourcie dans les souris transgéniques
PML-RARα en présence du produit de fusion réciproque RARα-PML, de FLT3-ITD ou d’une
hyper-expression de Bcl2 [33-35].
De plus, des anomalies chromosomiques secondaires sont détectées dans tous les
modèles murins (dont les plus fréquentes sont la trisomie 15, la trisomie 8 et des pertes des
chromosomes sexuels), ainsi que dans environ 40% des cas humains de LAP, trisomie 8

71
principalement [84], pouvant participer à la pathogenèse de la LAP.
La t(15 ;17) serait donc l’événement primaire initiant la leucémogenèse. L’acquisition
d’anomalies génétiques additionnelles coopérerait avec PML-RARα pour mener au phénotype
leucémique complet.
I-2-2. Quels mécanismes pourraient être à l’origine de l’acquisition d’évènements
secondaires ?
PML joue un rôle dans la stabilité génomique par son interaction avec la protéine
BLM. Le gène BLM code pour une hélicase de l’ADN de la famille RecQ. Il est muté dans le
syndrome de Bloom qui est caractérisé au niveau cellulaire par une instabilité génomique
entraînant des cassures chromosomiques et des échanges de chromatide excessifs [85]. La
susceptibilité excessive aux mutations et aux recombinaisons homologues des cellules des
patients atteints du syndrome de Bloom se traduit cliniquement par une prédisposition très
importante aux cancers. Le rôle précis de la protéine BLM dans la stabilité génomique est
encore imparfaitement connu : elle est impliquée dans la réparation de l’ADN lors de la
réplication [86] en interagissant avec p53 [87] et la protéine hMSH6 du système de réparation
des mésappariements [88]. La protéine BLM est localisée dans le noyau, dans les corps
nucléaires associés à PML (NBs) [89]. Il a été montré que PML permet de maintenir la
protéine BLM au sein des NBs, localisation nécessaire à sa fonction normale dans le maintien
de la stabilité génomique. Les NB étant désorganisés dans les cellules PML-RARα+, la
protéine BLM est délocalisée, il en résulte une augmentation des SCE (sister chromatin
exchange), témoins d’une instabilité génomique [90].
De plus, la perte de fonctionnalité de PML dans la LAP semble conduire à un défaut
d’apoptose qui permettrait le maintien de l’instabilité génomique. En effet, PML est un facteur
pro-apoptotique comme le montre la protection vis à vis des effets létaux des radiations
ionisantes et de l’anticorps anti-Fas des cellules PML-/- [91].
Cette instabilité génomique pourrait être due à la présence de la protéine de fusion
réciproque RARα-PML comme le suggèrent les travaux de Zimonjic et col.[92]. Cette équipe
a étudié la présence d’anomalies chromosomiques secondaires par analyse caryotypique
spectrale chez des souris transgéniques PML-RARα+ (PR+) ou PML-RARα+ et RARα-
PML+ (PR+RP+) leucémiques. Les souris PR+ ne présentent pas d’anomalie chromosomique

72
secondaire contrairement aux souris PR+RP+ qui acquièrent dans la majorité des cas (11/13)
une délétion du chromosome 2 récurrente, associée fréquemment à des anomalies de nombre
supplémentaires. Ces données suggèrent que l’expression de l’isoforme RARα-PML facilite
l’acquisition d’évènements génétiques secondaires procurant probablement un avantage
sélectif à la cellule leucémique.
Il semble donc exister un état d’instabilité génétique dans la LAP lié à la présence de la
t(15 ;17) qui pourrait être à l’origine de l’expression aberrante des gènes affiliés à la lignée
lymphoïde T dans les LAPv par dérégulation chromatinienne et qui pourrait aussi expliquer la
fréquence élevée des duplications du gène FLT3 dans cette pathologie.
I-3. Limites du modèle de l’infidélité de lignée
Dans un contexte d’instabilité génomique, la dérégulation de l’expression des gènes
n’obéit à aucune règle, elle se fait au hasard dans une cellule leucémique donnée et donc
différemment d’une cellule à l’autre. En conséquence, l’expression aberrante des gènes varie
d’une population blastique à une autre de façon aléatoire, sauf si elle apporte un avantage
prolifératif à la cellule et qu’elle est sélectionnée. Mais, l’acquisition aberrante de marqueurs
lymphoïdes T (pTα/TEA, réarrangements des TCR, CD2, cCD3, TdT) n’apporte à priori
aucun bénéfice de survie pour la cellule, les niveaux d’expression des gènes lymphoïdes T
devraient être aléatoires et se distribuer selon la loi normale. Or, nous avons observé une
expression de pTα/TEA significativement plus élevée dans les LAPv que dans les LAPc, ce
qui n’est pas cohérent avec une expression liée au hasard. La répression constante de
l’expression de RAG1 n’est pas non plus en faveur de cette hypothèse, car dans ce modèle,
l’expression aberrante concerne potentiellement tous les gènes, incluant RAG1.
II. PROMISCUITE DE LIGNEE
Les blastes leucémiques sont des cellules bloquées à un stade précoce de leur
maturation par le processus tumoral. Ils ont des capacités de prolifération limitées suggérant
qu’une sous-population leucémique possédant des capacités de prolifération importantes ainsi

73
que des propriétés d’autorenouvellement permet le maintien de la leucémie. Ces cellules sont
considérées comme des « cellules souches leucémiques » [93] dans lesquelles sont survenus
les évènements oncogéniques initiaux responsables de la transformation leucémique. Selon le
modèle de la promiscuité de lignée, la co-expression de marqueurs myéloïdes et lymphoïdes
serait le reflet du phénotype de la cellule souche leucémique perpétué dans les cellules
blastiques [94]. Ainsi, la présence de multiples caractéristiques d’orientation vers la lignée T
dans les LAPv serait en rapport avec la nature de la cellule cible de la t(15 ;17).
II-1. Légitimité du modèle de la promiscuité de lignée
Une étude récente visant à déterminer la cellule cible de la transformation leucémique
par les gènes de fusion impliquant MLL montre l’influence de la nature de la cellule cible sur
le phénotype de la cellule blastique et renforce l’hypothèse de la promiscuité de lignée [95].
MLL est un gène localisé en 11q23 impliqué dans des fusions avec de multiples gènes
partenaires. Les translocations impliquant MLL sont associées à des LAM, des LAL et des LA
biphénotypiques, c'est-à-dire co-exprimant des marqueurs myéloïdes et lymphoïdes. Dans
cette étude, les auteurs utilisent un modèle de cellules souches hématopoïétiques (CSH)
murines transduites par la fusion MLL-GAS7 et montrent que ces cellules induisent des LAM,
des LAL et LA biphénotypiques lorsqu’elles sont injectées à des souris irradiées. In vitro, la
transformation par MLL-GAS7 de populations cellulaires purifiées indique que seules les
CSH transformées produisent des colonies biphénotypiques en culture, tandis que les
progéniteurs myéloïdes et les progéniteurs lymphoïdes transformés induisent des colonies
uniquement myéloïdes ou lymphoïdes respectivement. Ces données suggèrent que les LA
biphénotypiques associées à MLL résultent des effets oncogéniques de MLL sur les
progéniteurs multipotents, confortant ainsi le modèle de la promiscuité de lignée.
En respectant cette hypothèse, l’expression de multiples caractéristiques
d’appartenance à la lignée lymphoïde T dans les LAPv est en faveur de la survenue de la
t(15 ;17) dans un progéniteur non engagé définitivement dans la lignée myéloïde, ayant
conservé un potentiel de différenciation vers la lignée T.

74
II-2. Comment replacer la cellule cible de la t(15 ;17) dans les LAPv
dans l’hématopoïèse à la lumière des marqueurs T retrouvés ?
II-2-1. Transformation d’un progéniteur avant l’engagement vers la lignée
myéloïde
L’expression concomitante de gènes affiliés à différentes lignées a été mise en
évidence dans les cellules souches et les progéniteurs avant leur orientation définitive vers une
lignée. Hu et col . ont montré par RT-PCR sur cellule unique la co-expression faible de gènes
myéloérythoïdes (incluant myélopéroxydase et β-globine) dans une cellule multipotente [47].
Récemment, une analyse du transcriptome par micro-array réalisée sur des populations
rigoureusement purifiées de CSH, progéniteurs multipotents (MPP), progéniteurs myéloïdes
communs (CMP) et progéniteurs lymphoïdes communs (CLP) a montré que les CSH co-
expriment de multiples gènes hématopoïétiques et non hématopoïétiques; les MPP co-
expriment des gènes lymphoïdes et myéloïdes; les CMP expriment des gènes myéloérythoïdes
mais pas de gènes lymphoïdes tandis que les CLP expriment des gènes lymphoïdes T, B et NK
mais pas myéloïdes [96]. Ces données suggèrent que les cellules souches présentent une
structure chromatinienne ouverte de tout le génome qui maintient leur pluripotentialité.
L’accessibilité transcriptionnelle est progressivement restreinte à mesure que la cellule se
différencie dans une lignée particulière. Ce concept est renforcé par des études menées sur le
facteur de transcription Pax5 impliqué dans la lymphopoïèse B précoce. Les cellules pro-B
Pax5-/- ne sont pas restreintes à la lignée B puisqu’elles peuvent se différencier en
macrophages, ostéoclastes, cellules dendritiques, granulocytes et cellules NK fonctionnels.
Elles expriment des gènes des différentes lignées, et la restauration de l’activité Pax5 réprime
cette transcription multi-lignée [97]. Ceci suggère que le processus d’engagement vers une
lignée est corrélé avec une extinction progressive des gènes des autres lignées.
La co-expression de marqueurs myéloïdes (CD13, CD33, MPO…) et de
caractéristiques d’orientation vers la lignée T (pTα/TEA, CD2, cCD3, TdT…) dans les LAPv
suggère donc que la cellule cible de la t(15 ;17) n’est pas encore engagée dans la lignée
myéloïde puis qu’elle n’a pas réprimé ses gènes lymphoïdes T.

75
II-2-2. Quel progéniteur non engagé définitivement dans la lignée myéloïde
pourrait être la cible de la t(15 ;17) ?
Selon le modèle de l’hématopoïèse défendu par Weissman (figure 4, haut) les CMP et
CLP ne peuvent être la cible de la t(15 ;17) puisqu’ils expriment des gènes uniquement
myéloïdes et lymphoïdes (T, B et NK) respectivement [96].
Selon le modèle alternatif proposé par Katsura (figure 4, bas), le progéniteur bipotent T/M
semble être un candidat intéressant pour expliquer la coexistence des caractéristiques
myéloïdes et T.
Cependant, dans la mesure où nous n’avons pas recherché de manière approfondie la
présence de marqueurs affiliés à la lignée B, nous ne pouvons exclure la possibilité de
l’implication du progéniteur multipotent M/T/B, voire de la CSH, bien que nous n’ayons
retrouvé CD19 que dans 15% des LAPv.
II-2-3. LAP microgranulaire et LAP classique: origine cellulaire différente ?
Si en respectant le modèle de la promiscuité de lignée, il apparaît que la transformation
en LAPv s’effectue dans un progéniteur primitif, non myéloïde restreint, il est légitime de
penser que la faible expression de marqueurs lymphoïdes T dans les LAPc est liée à une
origine cellulaire différente, au niveau d’un progéniteur engagé vers la lignée myéloïde.
Ainsi, les données contradictoires de la littérature concernant la cible de la transformation
leucémique peuvent être rediscutées.
Les travaux menés par Bonnet et Dick [38] visant à caractériser la cellule souche
leucémique en l’isolant d’une population blastique humaine par détermination de sa capacité à
induire un envahissement médullaire chez la souris NOD/SCID, sont en faveur de
l’implication d’un progéniteur engagé. En effet, dans cette étude, les blastes dérivant de 3
patients atteints de LAP (de type morphologique non précisé) sont incapables de coloniser la
moelle des souris NOD/SCID contrairement aux autres types de LAM. Cependant, d’autres
travaux [98] utilisant le même modèle retrouvent un envahissement médullaire pour 3 cas de
LAP sur 5 étudiés, bien que le nombre de cellules humaines à 8 semaines après la greffe soit
significativement plus bas par rapport aux autres types de LAM (1.8% versus 20.5%). Ces
données sont en faveur de la survenue de la transformation leucémique dans un progéniteur
primitif, pour au moins un sous-groupe de LAP. Les faibles niveaux d’envahissement

76
observés suggèrent que le pool de SL-IC (cellules souches leucémiques) est réduit dans la
LAP ou que leur capacité de croissance et de différenciation est plus faible que celle des
autres LAM. Le type morphologique de ces cas n’est malheureusement pas précisé.
Les autres travaux en faveur de la transformation d’un progéniteur engagé reposent sur la mise
en évidence de PML-RARα uniquement dans une population différenciée. Turhan et col. [40]
ont détecté l’expression de PML-RARα par RT-PCR dans 3 cas de LAP uniquement au sein
de la fraction CD34+/CD38+. A l’inverse, Takatsuki et col. [44] ont retrouvé la présence de
PML-RARα par RT-PCR dans les CFU-GM et les BFU-E de 2 cas sur 5 examinés, suggérant
que la t(15 ;17) survient dans une cellule souche pluripotente chez certains patients.
Malheureusement, aucune donnée concernant la morphologie et l’immunophénotype de ces
cas n’est précisée. Les données contradictoires de ces 2 études peuvent être liées au sous-type
de LAP analysées, (LAPc pour Turhan et LAPv pour Takatsuki), mais aussi à des différences
de sensibilité de la RT-PCR ou à un manque de pureté des populations étudiées.
Cependant, la mise en évidence de la fusion PML-RARα par FISH dans la majorité
des cellules de la fraction CD34+CD38- dans 2 cas de LAPv par Edwards et col. [43] est un
argument fort en faveur de la transformation d’un progéniteur non engagé dans les LAPv.
L’étude de Haferlach et col. [41] dans laquelle la fusion PML-RARα est recherchée par
FICTION (FISH et immunophénotypage) n’est pas en accord avec celle d’Edwards puisque
l’expression de PML-RARα est restreinte à la lignée myéloïde, mais il s’agissait de 6 cas de
LAP hypergranulaires.
Finalement, les données contradictoires des travaux visant à caractériser la cellule
souche leucémique dans la LAP peuvent être « réconciliées » si l’on considère que 2 cellules
de nature différente peuvent être la cible de la t(15 ;17) : l’une engagé vers la lignée myéloïde
(LAP hypergranulaire), l’autre primitive (LAP microgranulaire). Il est d’ailleurs intéressant de
noter que dans lorsque PML-RARα est exprimé dans des progéniteurs primitifs murin
CD34+Lin-, les souris développent une forme cytologique de LAP se rapprochant de la forme
variante humaine [32].
Il existe une association entre la morphologie variante, l’hyperleucocytose, la présence
de FLT3-ITD, et le variant moléculaire BCR3 (dans certaines séries). Ces autres différences
entre LAPv et LAPc pourraient peut-être elles aussi être rattachées à une origine cellulaire
différente. Il est possible que les différents progéniteurs permissifs pour la t(15 ;17) soient

77
susceptibles à différents types de lésions génétiques entraînant des caractéristiques
biologiques différentes. Cependant, nous n’avons pas retrouvé de corrélation entre les taux de
pTα/TEA et FLT3-ITD ou BCR3.
II-3. Interprétation hypothétique des niveaux de pTαααα/TEA dans les
LAM contrôles
Si la majorité des sous-types de LAM, en dehors des LAP, sont issus de la
transformation de progéniteurs multipotents CD34+/CD38- comme le montrent les travaux de
Bonnet et Dick, selon le concept de la promiscuité des lignées myéloïde et lymphoïde au
niveau des cellules souches par configuration chromatinienne ouverte de tout le génome, nous
devrions retrouver une faible expression de pTα/TEA dans l’ensemble des LAM contrôles.
Or, si les LAM 0/1/2 présentent un niveau faible de pTα/TEA, les LAM monocytaires 4/5
montrent un niveau indétectable de transcrits pTα/TEA. Ceci pourrait constituer un argument
supplémentaire en faveur de l’indépendance des voies de différenciation granuleux/T d’un
côté, et monocyte-macrophage/B de l’autre. En effet, il a été montré que les cellules B
pouvaient se différencier en macrophage [99] et il a été récemment mis en évidence un
progéniteur bipotent B/macrophage dans la moelle osseuse adulte. Ces progéniteurs
représentent environ 0.5% de la moelle osseuse totale et ne peuvent se différencier en
lymphocyte T ou NK [100].
II-4. Limites du modèle de la promiscuité de lignée
Le niveau d’expression de pTα retrouvé dans les LAPv est élevé, comparable à celui
des LAL-T pré-αβ qui présentent un phénotype de précurseur thymique exprimant un pré-
TCR, CD4/CD8 DP, ayant subi ou allant subir le processus de β-sélection. A ce stade, le
précurseur thymique est engagé définitivement dans la différenciation T, présente
constamment des réarrangements des TCR δ, γ ou β et a une expression forte de pTα corrélée
à celle de RAG1 [79]. Le niveau retrouvé dans les LAPv est donc celui d’un progéniteur T
engagé, ce qui est incohérent avec la négativité de RAG1 et l’absence de réarrangements des
TCR δ et γ dans 73% des cas. Le niveau d’expression de pTα attendu dans un précurseur bi ou
multipotent est faible.

78
De la même façon, l’expression de pTα dans les LAPc, bien que faible, est plus forte
que celle attendue pour un progéniteur engagé dans la différenciation granuleuse qui devrait
avoir réprimé complètement ses gènes lymphoïdes T.
Ces observations suggèrent qu’il y a probablement une part de dérégulation de
l’expression des gènes par la protéine de fusion PML-RARα, qui activerait le gène pTα.
III. CONCLUSION
Deux modèles s’opposent pour expliquer la présence de caractéristiques affiliés à la
lignée lymphoïde T. Selon le modèle de l’infidélité de lignée, il s’agit d’une expression
inapproprié, aberrante, résultant d’une dérégulation de l’expression des gènes dans un
contexte d’instabilité génomique lié à la présence de PML-RARα. Le modèle de la
promiscuité de lignée indique que la présence de caractéristiques lymphoïdes T serait le reflet
de la nature de la cellule souche leucémique, cible de la t(15 ;17).
Le modèle de l’infidélité nous semble insuffisant. Une action directe de PML-RARα
sur les séquences régulatrices des gènes lymphoïdes T paraît exclue devant l’absence
d’implication de la voie de l’acide rétinoïque dans la différenciation T ainsi que l’absence de
séquences RAREs dans le gène pTα. Si la stabilité génomique semble réellement altérée dans
la LAP, qu’une dérégulation chromatinienne soit responsable de l’expression de l’ensemble
des gènes T retrouvés nous paraît improbable. En effet, les différences observées entre les
LAPc et les LAPv ainsi que la répression constante du gène RAG1 n’est pas cohérent avec
une expression génique liée au hasard.
La multiplicité des caractéristiques d’orientation vers la lignée lymphoïde T rend plus
vraisemblable l’hypothèse d’une promiscuité de lignée. La présence des transcrits affiliés à la
lignée T ainsi que la présence de réarrangements du TCR dans la LAPv serait alors le reflet
des caractéristiques de la cellule souche leucémique perpétuées dans la descendance blastique,
indiquant que la t(15 ;17) surviendrait dans un progéniteur primitif, non myéloïde-restreint,
ayant un potentiel de différenciation T. Le concept récent selon lequel la cellule souche
hématopoïétique normale présente une conformation chromatinienne ouverte de tout le
génome avec expression faible de transcrits de toutes les lignées progressivement restreinte au
fur et à mesure de la différenciation est un argument fort en faveur de la survenue de la

79
t(15 ;17) dans un progéniteur primitif dans la LAPv. Ce progéniteur aurait donc un double
potentiel, myéloïde et T. Le meilleur candidat est le progéniteur bipotent myéloïde/T
récemment mis en évidence par les travaux de l’équipe de Katsura qui remettent en cause le
modèle de l’hématopoïèse communément admis reposant sur la séparation précoce entre les
lignées lymphoïdes et myéloïdes. Néanmoins, le niveau de pTα dans les LAPv est aussi élevé
que dans les précurseurs thymiques engagés au moment de la β-sélection suggérant tout de
même une part de dérégulation de l’expression de pTα.
Finalement, en tenant compte de toutes nos observations, nous pouvons représenter les
cellules cibles de la transformation leucémique présumées des différent sous-types de
leucémie aiguë de la manière suivante :
Figure 18. Représentation hypothétique des progéniteurs cibles de la transformation leucémique dans les différents sous-types de leucémies aiguës, selon le modèle de l’hématopoïèse proposé par Katsura
�
CSH EMeg M/T/B
CMLP
M/T
Mφφφφ/B
T
M
Mφφφφ
�
LAPc
LAL-T
LAM 4/5
LAPv
LAM 0/1/2
B

80
CONCLUSION ET PERSPECTIVES
Nous avons mis en évidence dans les formes variantes de LAP l’expression des
transcrits pTα et TEA, en association à d’autres marqueurs lymphoïde T, comme l’expression
de CD2, cCD3 ou TdT et la présence de réarrangements du TCR γ et δ.
Ces résultats indiquent que les blastes de LAP variante présentent des caractéristiques
multiples d’orientation vers la lignée T, reflétant soit une dérégulation de l’expression des
gènes liée au processus leucémique, soit plus probablement l’immuno-génotype de la cellule
souche leucémique cible de la transformation leucémique. Ces données suggèrent donc que,
dans les LAP variantes, la t(15 ;17) surviendrait dans un progéniteur plus primitif non
myéloïde restreint ayant conservé un potentiel de différenciation vers la lignée T confortant
ainsi l’existence d’un progéniteur bipotent myéloïde/T récemment mis en évidence.
Afin de comprendre de façon plus fine et plus large dans quelle mesure les blastes de
LAP sont apparentés à la lignée lymphoïde T, nous envisageons de réaliser une analyse du
profil d’expression génique sur puces (micro-array) dans laquelle nous comparerons le
transcriptome des formes classiques de LAP à celui des formes microgranulaires. Ainsi, nous
pourrons étudier l’expression de plus nombreux transcrits lymphoïdes T et évaluer le statut
des transcrits des autres lignées, en particulier la lignée B. Nous espérons aussi confirmer les
différences d’expression des transcrits T entre les deux formes de LAP pour conforter l’idée
qu’elles dérivent de deux progéniteurs différents.
�

81
BIBLIOGRAPHIE
1. Bennett, J.M., et al., Proposals for the classification of the acute leukaemias. French-
American-British (FAB) co-operative group. Br J Haematol, 1976. 33(4): p. 451-8. 2. Grimwade, D., et al., The importance of diagnostic cytogenetics on outcome in AML:
analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood, 1998. 92(7): p. 2322-33.
3. Harris, N.L., et al., The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the Clinical Advisory Committee meeting, Airlie House, Virginia, November, 1997. Ann Oncol, 1999. 10(12): p. 1419-32.
4. Douer, D., The epidemiology of acute promyelocytic leukaemia. Best Pract Res Clin Haematol, 2003. 16(3): p. 357-67.
5. Fenaux, P., et al., A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood, 1999. 94(4): p. 1192-200.
6. De Rossi, G., et al., Immunological definition of acute promyelocytic leukemia (FAB M3): a study of 39 cases. Eur J Haematol, 1990. 45(3): p. 168-71.
7. Claxton, D.F., et al., Correlation of CD2 expression with PML gene breakpoints in patients with acute promyelocytic leukemia. Blood, 1992. 80(3): p. 582-6.
8. Biondi, A., et al., CD2 expression in acute promyelocytic leukemia is associated with microgranular morphology (FAB M3v) but not with any PML gene breakpoint. Leukemia, 1995. 9(9): p. 1461-6.
9. Guglielmi, C., et al., Immunophenotype of adult and childhood acute promyelocytic leukaemia: correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br J Haematol, 1998. 102(4): p. 1035-41.
10. Paietta, E., et al., A surrogate marker profile for PML/RAR alpha expressing acute promyelocytic leukemia and the association of immunophenotypic markers with morphologic and molecular subtypes. Cytometry, 2004. 59B(1): p. 1-9.
11. Grimwade, D., et al., Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Francais de Cytogenetique Hematologique, Groupe de Francais d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood, 2000. 96(4): p. 1297-308.
12. Chen, Z., et al., Fusion between a novel Kruppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia. Embo J, 1993. 12(3): p. 1161-7.
13. Wells, R.A., C. Catzavelos, and S. Kamel-Reid, Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet, 1997. 17(1): p. 109-13.

82
14. Redner, R.L., et al., The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood, 1996. 87(3): p. 882-6.
15. Arnould, C., et al., The signal transducer and activator of transcription STAT5b gene is a new partner of retinoic acid receptor alpha in acute promyelocytic-like leukaemia. Hum Mol Genet, 1999. 8(9): p. 1741-9.
16. Mandelli, F., et al., Molecular remission in PML/RAR alpha-positive acute promyelocytic leukemia by combined all-trans retinoic acid and idarubicin (AIDA) therapy. Gruppo Italiano-Malattie Ematologiche Maligne dell'Adulto and Associazione Italiana di Ematologia ed Oncologia Pediatrica Cooperative Groups. Blood, 1997. 90(3): p. 1014-21.
17. Rowley, J.D., H.M. Golomb, and C. Dougherty, 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet, 1977. 1(8010): p. 549-50.
18. de The, H., et al., The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell, 1991. 66(4): p. 675-84.
19. Mistry, A.R., et al., The molecular pathogenesis of acute promyelocytic leukaemia: implications for the clinical management of the disease. Blood Rev, 2003. 17(2): p. 71-97.
20. Collins, S.J., The role of retinoids and retinoic acid receptors in normal hematopoiesis. Leukemia, 2002. 16(10): p. 1896-905.
21. Zhong, S., P. Salomoni, and P.P. Pandolfi, The transcriptional role of PML and the nuclear body. Nat Cell Biol, 2000. 2(5): p. E85-90.
22. Ishov, A.M., et al., PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol, 1999. 147(2): p. 221-234.
23. Strudwick, S. and K.L. Borden, Finding a role for PML in APL pathogenesis: a critical assessment of potential PML activities. Leukemia, 2002. 16(10): p. 1906-17.
24. Wang, Z.G., et al., Role of PML in cell growth and the retinoic acid pathway. Science, 1998. 279(5356): p. 1547-51.
25. Ruggero, D., Z.G. Wang, and P.P. Pandolfi, The puzzling multiple lives of PML and its role in the genesis of cancer. Bioessays, 2000. 22(9): p. 827-35.
26. Di Croce, L., et al., Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science, 2002. 295(5557): p. 1079-82.
27. Grignani, F., et al., The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell, 1993. 74(3): p. 423-31.
28. Grignani, F., et al., PML/RAR alpha fusion protein expression in normal human hematopoietic progenitors dictates myeloid commitment and the promyelocytic phenotype. Blood, 2000. 96(4): p. 1531-7.
29. Grisolano, J.L., et al., Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood, 1997. 89(2): p. 376-87.
30. Brown, D., et al., A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci U S A, 1997. 94(6): p. 2551-6.
31. He, L.Z., et al., Acute leukemia with promyelocytic features in PML/RARalpha transgenic mice. Proc Natl Acad Sci U S A, 1997. 94(10): p. 5302-7.
32. Minucci, S., et al., PML-RAR induces promyelocytic leukemias with high efficiency

83
following retroviral gene transfer into purified murine hematopoietic progenitors. Blood, 2002. 100(8): p. 2989-95.
33. Pollock, J.L., et al., A bcr-3 isoform of RARalpha-PML potentiates the development of PML-RARalpha-driven acute promyelocytic leukemia. Proc Natl Acad Sci U S A, 1999. 96(26): p. 15103-8.
34. Kelly, L.M., et al., PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc Natl Acad Sci U S A, 2002. 99(12): p. 8283-8.
35. Kogan, S.C., et al., BCL-2 cooperates with promyelocytic leukemia retinoic acid receptor alpha chimeric protein (PMLRARalpha) to block neutrophil differentiation and initiate acute leukemia. J Exp Med, 2001. 193(4): p. 531-43.
36. Park, C.H., D.E. Bergsagel, and E.A. McCulloch, Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst, 1971. 46(2): p. 411-22.
37. Bergsagel, D.E. and F.A. Valeriote, Growth characteristics of a mouse plasma cell tumor. Cancer Res, 1968. 28(11): p. 2187-96.
38. Bonnet, D. and J.E. Dick, Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med, 1997. 3(7): p. 730-7.
39. Passegue, E., et al., Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci U S A, 2003. 100 Suppl 1: p. 11842-9.
40. Turhan, A.G., et al., Highly purified primitive hematopoietic stem cells are PML-RARA negative and generate nonclonal progenitors in acute promyelocytic leukemia. Blood, 1995. 85(8): p. 2154-61.
41. Haferlach, T., et al., Cell lineage specific involvement in acute promyelocytic leukaemia (APL) using a combination of May-Grunwald-Giemsa staining and fluorescence in situ hybridization techniques for the detection of the translocation t(15;17)(q22;q12). Br J Haematol, 1998. 103(1): p. 93-9.
42. Vickers, M., G. Jackson, and P. Taylor, The incidence of acute promyelocytic leukemia appears constant over most of a human lifespan, implying only one rate limiting mutation. Leukemia, 2000. 14(4): p. 722-6.
43. Edwards, R.H., et al., Evidence for early hematopoietic progenitor cell involvement in acute promyelocytic leukemia. Am J Clin Pathol, 1999. 112(6): p. 819-27.
44. Takatsuki, H., et al., PML/RAR alpha fusion gene is expressed in both granuloid/macrophage and erythroid colonies in acute promyelocytic leukaemia. Br J Haematol, 1993. 85(3): p. 477-82.
45. Grimwade, D., et al., The T-lineage-affiliated CD2 gene lies within an open chromatin environment in acute promyelocytic leukemia cells. Cancer Res, 2002. 62(16): p. 4730-5.
46. Jimenez, G., et al., Activation of the beta-globin locus control region precedes commitment to the erythroid lineage. Proc Natl Acad Sci U S A, 1992. 89(22): p. 10618-22.
47. Hu, M., et al., Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev, 1997. 11(6): p. 774-85.
48. Kondo, M., I.L. Weissman, and K. Akashi, Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell, 1997. 91(5): p. 661-72.
49. Akashi, K., et al., A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature, 2000. 404(6774): p. 193-7.
50. Katsura, Y., Redefinition of lymphoid progenitors. Nat Rev Immunol, 2002. 2(2): p. 127-32.
51. Kawamoto, H., et al., Emergence of T cell progenitors without B cell or myeloid

84
differentiation potential at the earliest stage of hematopoiesis in the murine fetal liver. J Immunol, 1999. 162(5): p. 2725-31.
52. Kawamoto, H., et al., T cell progenitors emerge earlier than B cell progenitors in the murine fetal liver. Immunity, 2000. 12(4): p. 441-50.
53. Lu, M., et al., The common myelolymphoid progenitor: a key intermediate stage in hemopoiesis generating T and B cells. J Immunol, 2002. 169(7): p. 3519-25.
54. Haddad, R., et al., Molecular characterization of early human T/NK and B lymphoid progenitor cells in umbilical cord blood. Blood, 2004.
55. Spits, H., et al., Early stages in the development of human T, natural killer and thymic dendritic cells. Immunol Rev, 1998. 165: p. 75-86.
56. Blom, B., et al., TCR gene rearrangements and expression of the pre-T cell receptor complex during human T-cell differentiation. Blood, 1999. 93(9): p. 3033-43.
57. von Boehmer, H., et al., Crucial function of the pre-T-cell receptor (TCR) in TCR beta selection, TCR beta allelic exclusion and alpha beta versus gamma delta lineage commitment. Immunol Rev, 1998. 165: p. 111-9.
58. von Boehmer, H., et al., Pleiotropic changes controlled by the pre-T-cell receptor. Curr Opin Immunol, 1999. 11(2): p. 135-42.
59. Khor, B. and B.P. Sleckman, Allelic exclusion at the TCRbeta locus. Curr Opin Immunol, 2002. 14(2): p. 230-4.
60. Del Porto, P., et al., Cloning and comparative analysis of the human pre-T-cell receptor alpha-chain gene. Proc Natl Acad Sci U S A, 1995. 92(26): p. 12105-9.
61. Ramiro, A.R., et al., Regulation of pre-T cell receptor (pT alpha-TCR beta) gene expression during human thymic development. J Exp Med, 1996. 184(2): p. 519-30.
62. Bruno, L., et al., Identification of a committed T cell precursor population in adult human peripheral blood. J Exp Med, 1997. 185(5): p. 875-84.
63. Collins, C., et al., RAG1, RAG2 and pre-T cell receptor alpha chain expression by adult human hepatic T cells: evidence for extrathymic T cell maturation. Eur J Immunol, 1996. 26(12): p. 3114-8.
64. Fehling, H.J., et al., Crucial role of the pre-T-cell receptor alpha gene in development of alpha beta but not gamma delta T cells. Nature, 1995. 375(6534): p. 795-8.
65. von Boehmer, H. and H.J. Fehling, Structure and function of the pre-T cell receptor. Annu Rev Immunol, 1997. 15: p. 433-52.
66. Kaufmann, S.H., gamma/delta and other unconventional T lymphocytes: what do they see and what do they do? Proc Natl Acad Sci U S A, 1996. 93(6): p. 2272-9.
67. Krangel, M.S., et al., A distinct wave of human T cell receptor gamma/delta lymphocytes in the early fetal thymus: evidence for controlled gene rearrangement and cytokine production. J Exp Med, 1990. 172(3): p. 847-59.
68. Bassing, C.H., W. Swat, and F.W. Alt, The mechanism and regulation of chromosomal V(D)J recombination. Cell, 2002. 109 Suppl: p. S45-55.
69. Mombaerts, P., et al., RAG-1-deficient mice have no mature B and T lymphocytes. Cell, 1992. 68(5): p. 869-77.
70. Wilson, A., W. Held, and H.R. MacDonald, Two waves of recombinase gene expression in developing thymocytes. J Exp Med, 1994. 179(4): p. 1355-60.
71. Langerak, A.W., et al., Basic helix-loop-helix proteins E2A and HEB induce immature T-cell receptor rearrangements in nonlymphoid cells. Blood, 2001. 98(8): p. 2456-65.
72. Lauzurica, P. and M.S. Krangel, Temporal and lineage-specific control of T cell receptor alpha/delta gene rearrangement by T cell receptor alpha and delta enhancers. J Exp Med, 1994. 179(6): p. 1913-21.
73. Biondi, A., et al., High prevalence of T-cell receptor V delta 2-(D)-D delta 3 or D

85
delta 1/2-D delta 3 rearrangements in B-precursor acute lymphoblastic leukemias. Blood, 1990. 75(9): p. 1834-40.
74. Shimizu, T., et al., Mouse germline transcript of TCR alpha joining region and temporal expression in ontogeny. Int Immunol, 1993. 5(2): p. 155-60.
75. de Chasseval, R. and J.P. de Villartay, Functional characterization of the promoter for the human germ-line T cell receptor J alpha (TEA) transcript. Eur J Immunol, 1993. 23(6): p. 1294-8.
76. Wilson, A., J.P. de Villartay, and H.R. MacDonald, T cell receptor delta gene rearrangement and T early alpha (TEA) expression in immature alpha beta lineage thymocytes: implications for alpha beta/gamma delta lineage commitment. Immunity, 1996. 4(1): p. 37-45.
77. Villey, I., et al., Defect in rearrangement of the most 5' TCR-J alpha following targeted deletion of T early alpha (TEA): implications for TCR alpha locus accessibility. Immunity, 1996. 5(4): p. 331-42.
78. Mauvieux, L., I. Villey, and J.P. de Villartay, T early alpha (TEA) regulates initial TCRVAJA rearrangements and leads to TCRJA coincidence. Eur J Immunol, 2001. 31(7): p. 2080-6.
79. Asnafi, V., et al., Analysis of TCR, pT alpha, and RAG-1 in T-acute lymphoblastic leukemias improves understanding of early human T-lymphoid lineage commitment. Blood, 2003. 101(7): p. 2693-703.
80. van Dongen, J.J., et al., Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia, 2003. 17(12): p. 2257-317.
81. Libura, M., et al., FLT3 and MLL intragenic abnormalities in AML reflect a common category of genotoxic stress. Blood, 2003. 102(6): p. 2198-204.
82. Dupret, C., et al., AMLs with MLL translocation can be separated into those with T and B lymphoid potentiel on the basis of their TCR and IgH genetic profiles. ASH oral session, 2003.
83. Smith, L.J., et al., Lineage infidelity in acute leukemia. Blood, 1983. 61(6): p. 1138-45.
84. Le Beau, M.M., et al., Recurring chromosomal abnormalities in leukemia in PML-RARA transgenic mice parallel human acute promyelocytic leukemia. Blood, 2002. 99(8): p. 2985-91.
85. Ellis, N.A., et al., The Bloom's syndrome gene product is homologous to RecQ helicases. Cell, 1995. 83(4): p. 655-66.
86. Adams, M.D., M. McVey, and J.J. Sekelsky, Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science, 2003. 299(5604): p. 265-7.
87. Sengupta, S., et al., BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. Embo J, 2003. 22(5): p. 1210-22.
88. Pedrazzi, G., et al., The Bloom's syndrome helicase interacts directly with the human DNA mismatch repair protein hMSH6. Biol Chem, 2003. 384(8): p. 1155-64.
89. Yankiwski, V., et al., Nuclear structure in normal and Bloom syndrome cells. Proc Natl Acad Sci U S A, 2000. 97(10): p. 5214-9.
90. Zhong, S., et al., A role for PML and the nuclear body in genomic stability. Oncogene, 1999. 18(56): p. 7941-7.
91. Wang, Z.G., et al., PML is essential for multiple apoptotic pathways. Nat Genet, 1998. 20(3): p. 266-72.

86
92. Zimonjic, D.B., et al., Acquired, nonrandom chromosomal abnormalities associated with the development of acute promyelocytic leukemia in transgenic mice. Proc Natl Acad Sci U S A, 2000. 97(24): p. 13306-11.
93. McCulloch, E.A., Stem cells in normal and leukemic hemopoiesis (Henry Stratton Lecture, 1982). Blood, 1983. 62(1): p. 1-13.
94. Greaves, M.F., et al., Lineage promiscuity in hemopoietic differentiation and leukemia. Blood, 1986. 67(1): p. 1-11.
95. So, C.W., et al., MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell, 2003. 3(2): p. 161-71.
96. Akashi, K., et al., Transcriptional accessibility for genes of multiple tissues and hematopoietic lineages is hierarchically controlled during early hematopoiesis. Blood, 2003. 101(2): p. 383-9.
97. Nutt, S.L., et al., Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature, 1999. 401(6753): p. 556-62.
98. Ailles, L.E., et al., Growth characteristics of acute myelogenous leukemia progenitors that initiate malignant hematopoiesis in nonobese diabetic/severe combined immunodeficient mice. Blood, 1999. 94(5): p. 1761-72.
99. Borrello, M.A., J. Palis, and R.P. Phipps, The relationship of CD5+ B lymphocytes to macrophages: insights from normal biphenotypic B/macrophage cells. Int Rev Immunol, 2001. 20(1): p. 137-55.
100. Montecino-Rodriguez, E., H. Leathers, and K. Dorshkind, Bipotential B-macrophage progenitors are present in adult bone marrow. Nat Immunol, 2001. 2(1): p. 83-8.

87
2004 ELISE CHAPIRO PRESIDENT DE THESE : Professeur Elizabeth Macintyre DIRECTEUR DE THESE : Docteur Vahid Asnafi
EXPRESSION DE TRANSCRITS AFFILIES A LA LIGNEE LYMPHOIDE T ET PRESENCE DE REARRANGEMENT DES TCR DANS LA LEUCEMIE AIGUE PROMYELOCYTAIRE :
IMPLICATIONS POUR LA CIBLE CELLULAIRE DE LA t(15 ;17)
RESUME La leucémie aiguë promyélocytaire (LAP) est communément considérée comme la forme la plus différenciée des leucémies aiguës myéloblastiques (LAM), dérivant d’un progéniteur restreint à la lignée myéloïde. Cependant, cette hypothèse est difficilement compatible avec l’observation qu’approximativement 25% des cas de LAP, particulièrement ceux avec la forme variante hypogranulaire (LAPv), expriment le marqueur lymphocytaire T-spécifique CD2. Afin de comprendre dans quelle mesure les cellules blastiques de LAP sont apparentées à la lignée lymphoïde T, nous avons étudié l’expression de transcrits associés aux étapes précoces de la différenciation lymphocytaire T (pré-T alpha, pTα, T early alpha, TEA et RAG1) par PCR quantitative et recherché des réarrangements des TCR dans une série de 36 cas de LAP avec t(15 ;17), incluant 21 cas de morphologie classique et 15 de forme variante. Nos résultats montrent que les 2 formes de LAP expriment pTα et TEA à des niveaux supérieurs à ceux de 26 cas de LAM d’autres types, avec des niveaux dans les LAPv comparables à ceux retrouvés dans les leucémies aiguës lymphoblastiques. Cependant aucune des 2 formes de LAP n’exprime RAG1. Les réarrangements des TCR sont plus fréquents dans les M3v. Ces résultats indiquent que les blastes de LAP variante présentent des caractéristiques multiples d’orientation vers la lignée T, reflétant soit une dérégulation de l’expression des gènes liée au processus leucémique, soit plus probablement l’immuno-génotype du progéniteur cible de la transformation leucémique. Ces données suggèrent donc que, dans les LAP variantes, la t(15 ;17) surviendrait dans un progéniteur plus primitif non myéloïde restreint.
MOTS-CLES :
-leucémie aiguë promyélocytaire
-hématopoïèse
-lymphocytes T
FACULTE DE MEDECINE DE CRETEIL 8 rue du Général Sarrail 94010 CRETEIL

88

89
EXPRESSION DE TRANSCRITS AFFILIES A LA LIGNEE LYMPHOIDE T ET PRESENCE DE REARRANGEMENTS DES TCR DANS LA LEUCEMIE AIGUE PROMYELOCYTAIRE : IMPLICATIONS POUR LA CIBLE CELLULAIRE DE LA t(15 ;17)
Résumé en français : La leucémie aiguë promyélocytaire (LAP) avec t(15 ;17) est considérée comme la plus différenciée des leucémies aiguës myéloblastiques, dérivant d’un progéniteur engagé dans la lignée myéloïde. Cependant, 25% des cas, particulièrement les formes variantes (M3v), expriment CD2, marqueur lymphoïde T spécifique. Pour comprendre dans quelle mesure les blastes de LAP sont apparentés à la lignée lymphoïde T, nous avons étudié l’expression de transcrits associés aux stades précoces de l’ontogénie T (pTα, TEA et RAG1) et recherché des réarrangements des TCR dans 21 LAP classiques et 15 M3v. Les 2 formes de LAP, mais particulièrement les M3v, expriment pTα et TEA à des niveaux élevés et présentent des réarrangements des TCR γ et δ. La présence de multiples caractéristiques de la lignée T dans les M3v reflète soit une dérégulation de l’expression des gènes liée au processus leucémique, soit plus probablement l’immunogénotype du progéniteur cible de la t(15 ;17) qui serait non myéloïde restreint. Résumé en anglais : Acute promyelocytic leukemia (APL) with t(15;17) is generally considered to be the most
differentiated acute myeloid leukemia deriving from late myeloid-restricted progenitors.
However, 25% of APL cases, particularly those with the hypogranular variant (M3v) form,
express the T-lineage affiliated marker, CD2. To gain further insight into the extent of T-
lineage deregulation in APL blasts, we quantified the expression of pre-Tα, TEA and RAG1
transcripts and determined TCR δ and γ status in 21 classical and 15 M3v APL cases. Both
classical and M3v APL, but particularly M3v, express pTα and TEA at high levels and have
TCR rearrangements. These data demonstrate that M3v exhibit several features suggestive of
early T-lymphoid orientation which may reflect either an aberrant gene expression coincident
with leukemogenesis or more probably the progenitor subject to leukemic transformation.
Thus, the t(15;17) may arise in more primitive progenitors in M3v than previously thought to
be the case.
Mots Clés : -Leucémie aiguë promyélocytaire -Hématopoïèse -Lymphocytes T

















![congresogto.s3.amazonaws.com · ep oseoo]d pp sopeunsaJ ap un ua 'sepasn e 01!tu9J '01en!eueno ap opelsa lap ose'ôuoo lap J0!Jedns ep oue6Jó lap 01uel-uelôe8 lap 'A uppoeJJ 9 'K](https://static.fdocuments.es/doc/165x107/5e9a40ae3586c73e69135011/congresogtos3-ep-oseood-pp-sopeunsaj-ap-un-ua-sepasn-e-01tu9j-01eneueno-ap.jpg)
