
CIRUGIA DE LAS FRACTURAS DE CRANEO TRAUMATISMO CRANEOENCEFALICO VII.
Upload
aryd87Category
view
38download
0
TCE: Fisiología, Fisiopatología, lesión primaria, lesión secundaria
Dra. Ariadna Muñoz Rojas RIUM

Anatomía y Fisiología

TENTORIO
Tienda del cerebelo es una división anatómica:
Supratentorial: fosa anterior y media.
Infratentorial: fosa posterior
Por la incisura tentorial o foramen magno. Pasa el tallo cerebral, por arriba está el uncus. Pasa III par craneal

FISIOLOGIA DEL SISTEMA NERVIOSO CENTRAL
Los nutrientes principales del cerebro son:
El oxígeno
Glucosa.
El cerebro es el tejido con menor tolerancia a la isquemia.
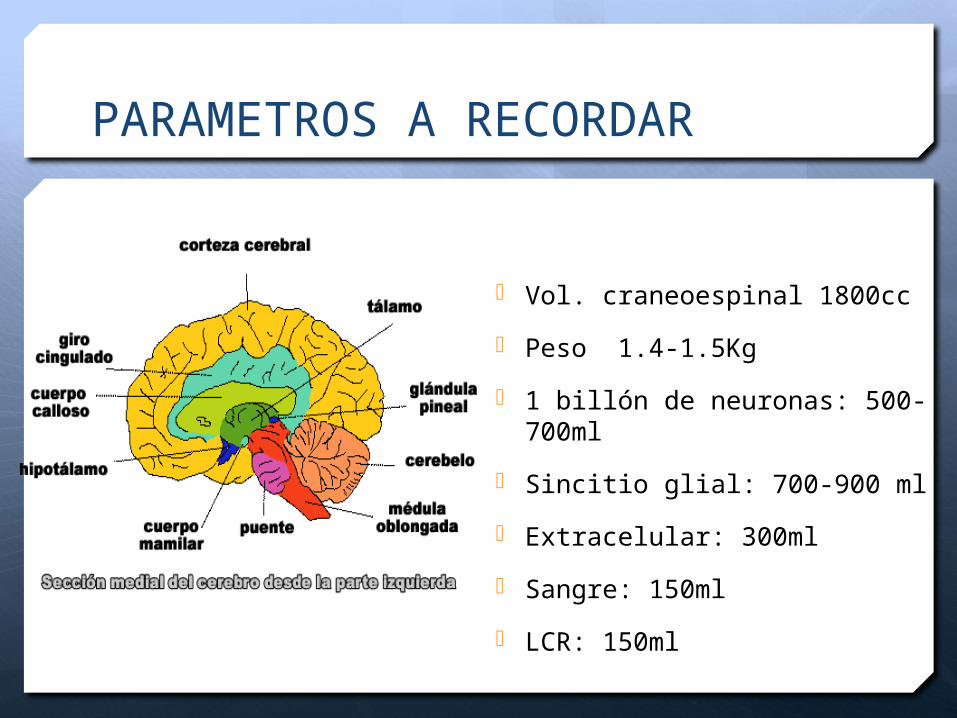
PARAMETROS A RECORDAR
Vol. craneoespinal 1800cc
Peso 1.4-1.5Kg
1 billón de neuronas: 500-700ml
Sincitio glial: 700-900 ml
Extracelular: 300ml
Sangre: 150ml
LCR: 150ml

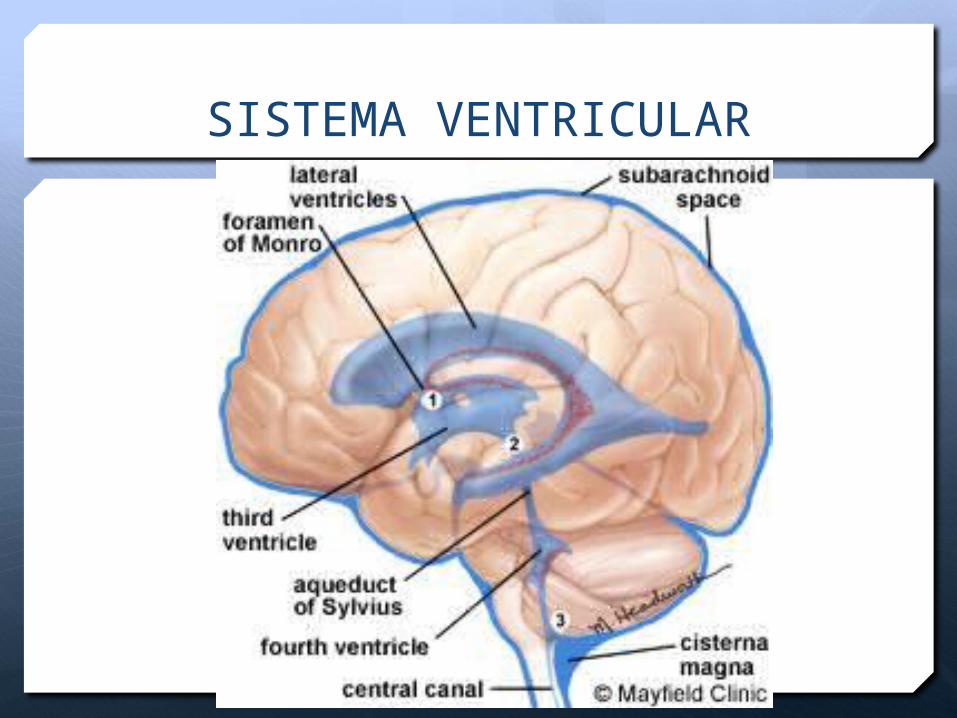
FISIOLOGÍA DEL LCR
LCR 150 ml total
40ml ventricular 80 ml espinal
70% 20%
30 ml subaracnoideo
10%
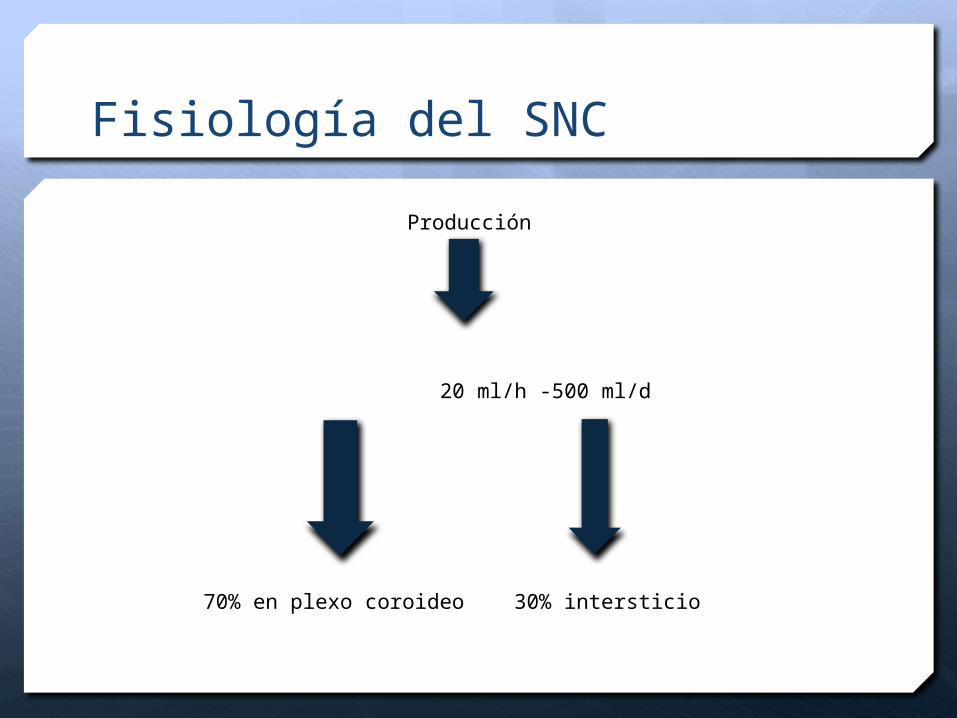
Fisiología del SNC
Producción
20 ml/h -500 ml/d
70% en plexo coroideo 30% intersticio

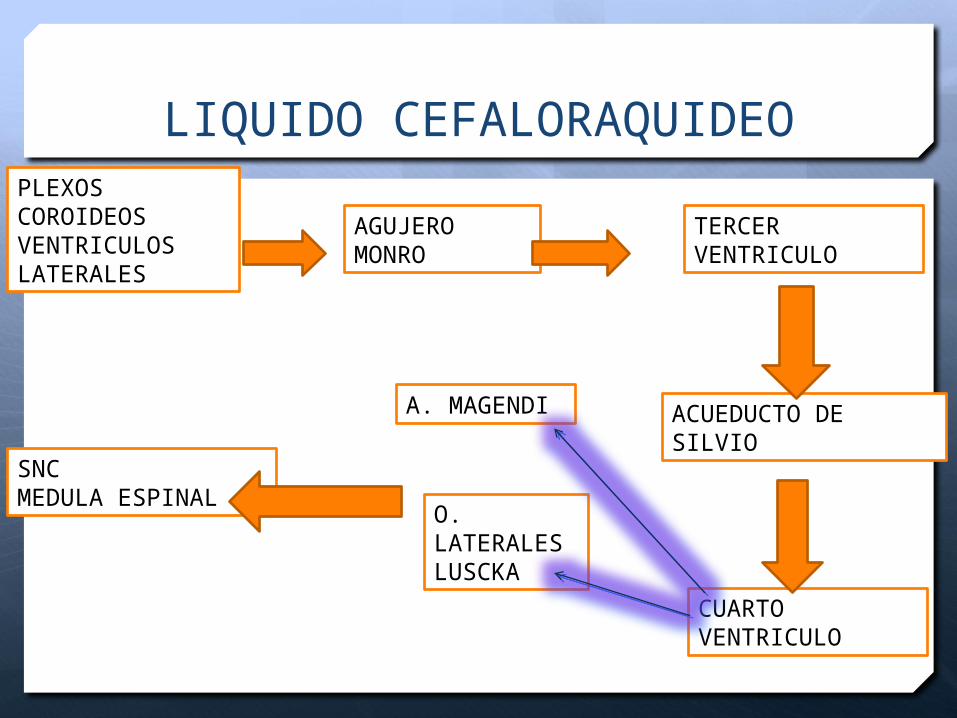
LIQUIDO CEFALORRAQUIDEO
Es producido por los plexos coroideos de los ventrículos (95%), así como por el epitelio ependimario.
La producción es de 0.3 ml/min (±450 ml/día), lo cual indica que el LCR se recambia hipotéticamente 3 veces al día.
LIQUIDO CEFALORAQUIDEO
PLEXOS COROIDEOSVENTRICULOS LATERALES
TERCER VENTRICULO
AGUJERO MONRO
ACUEDUCTO DE SILVIO
CUARTO VENTRICULO
A. MAGENDI
O. LATERALES LUSCKA
SNCMEDULA ESPINAL
SISTEMA VENTRICULAR
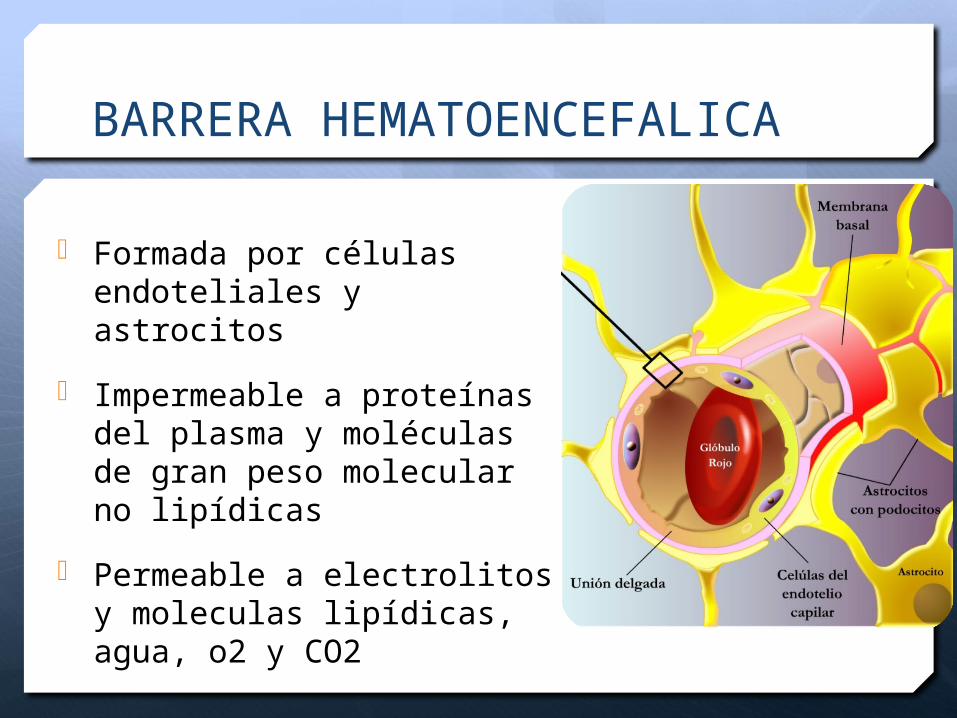
BARRERA HEMATOENCEFALICA
Formada por células endoteliales y astrocitos
Impermeable a proteínas del plasma y moléculas de gran peso molecular no lipídicas
Permeable a electrolitos y moleculas lipídicas, agua, o2 y CO2

CONSUMO DE OXIGENO
La oclusión del flujo mayor a 10 s disminuye la PaO2 rápidamente a 30 mmHg llevando al paciente a inconciencia.
15 seg tiene alteraciones en el EEG.
3 - 8 minutos se agotan las reservas de ATP
Lesión neuronal irreversible entre los 10 y 30 min siguientes.

METABOLISMO
HIPOTERMIA: afecta las reacciones bioquímicas
x 1 ºC disminuye el 7% consumo metabólico de O2 .
40-42ºC el CMRO aumenta 50% por cada ºC

FLUJO SANGUINEO
El flujo sanguíneo cerebral (FSC) normal es de 50-60 ml/ 100 g/min (750 ml/min) cuando la PCO2 es de 40 mmHG
Una partícula tarda 7 segundos desde la carótida interna hasta la yugular interna.
Si el FSC está entre 25 y 40 ml/100 g/min habrá disminución de la conciencia y menores de10 ml/100 g/min habrá muerte celular.

Teorías Regulación Flujo Sanguíneo Cerebral
Metabolica local
Miogénica: musculo liso vascular tiene la capacidad intrínse de detectar cambios de en la PPC por canales de calcio sensibles
Neurogénica: control por nervios perivasculares
Endotelio: factores endoteliales

FLUJO SANGUINEO CEREBRAL
Regulado por el CO2 es lineal mientras se mantenga en un rango de 25-60mmHg
Hipercapnea : vasodilatación
Aumentos mayores de 300mmHg de O2 causan vasoconstricción cerebral
Vasodilatación inicia con valores de O2 de 60mmHg

PRESION INTRACRANEAL
La presión intracraneana (PIC) normal en adultos es:
1 - 15 mmHg (50-180 mm de H2O)
Niños entre 1.5 a 7 mmHg;

PRESION DE PERFUSION ENCEFALICA (PPC)
Determinante principal de la presión o flujo sanguíneo encefálico
PPC = FSC /RVC
Parámetro regional circulatorio
Diferente a la presión arterial
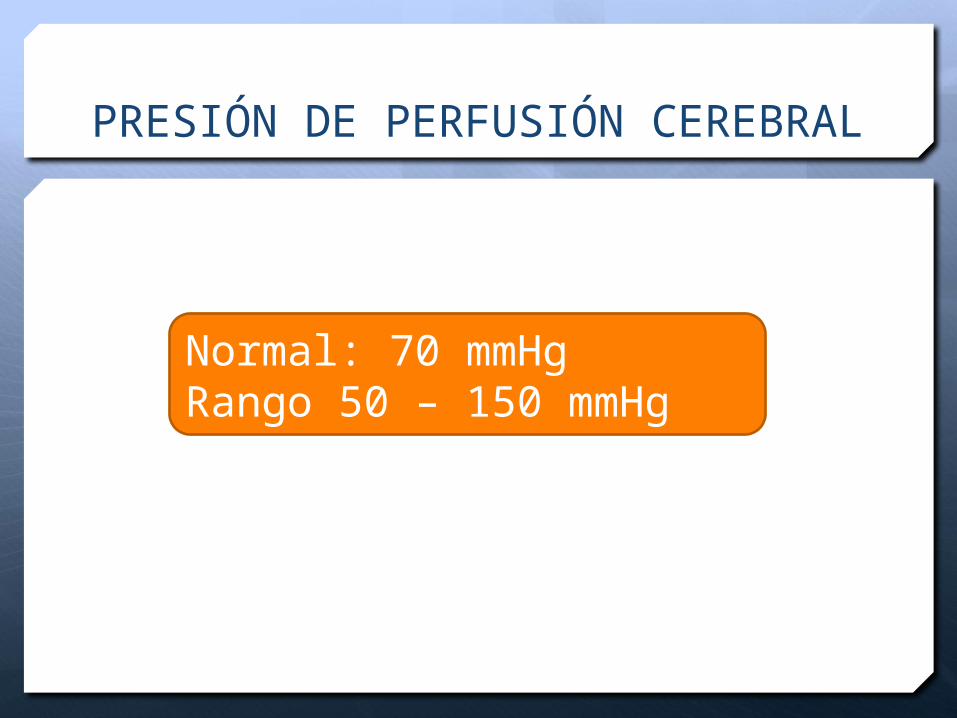
PRESIÓN DE PERFUSIÓN CEREBRAL
Normal: 70 mmHgRango 50 – 150 mmHg
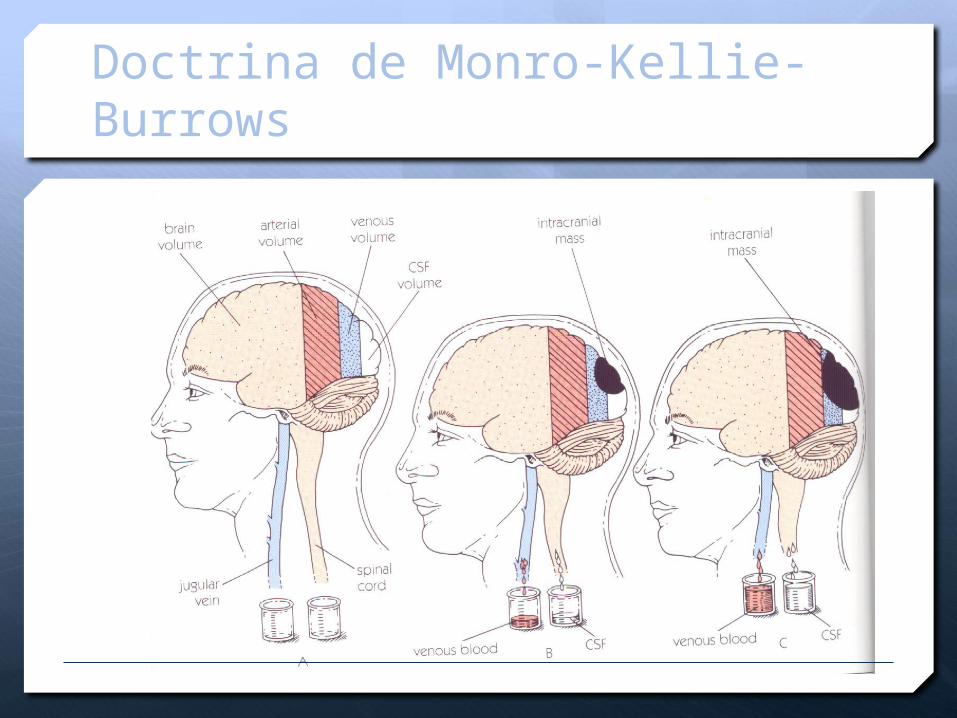
Doctrina de Monro-Kellie-Burrows

MONROE - KELLY
La cavidad intracraneana es un continente rígido y hermético compuesto por tres contenidos principales:
Parénquima intracraneano 80-85%
Líquido cefalorraquídeo 7.5-10%
Volumen sanguíneo 7.5-10%. (70% venoso, 30%
arterial)

DOCTRINA MONRO-KELLIE
Valor del volumen intracraneal = constante
Volumen intracraneal = V cerebro + V líquido cefalorraquídeo +V sanguíneo

MONROE KELLY
En caso de haber un crecimiento a través de semanas o meses de uno de estos contenidos, los demás se amoldarían en tamaño proporcional hasta cierto límite.
Esto no sucede en el trauma donde se tiene condiciones de aumento agudo de estos contenidos

MONROE KELLY
Parénquima intracraneano: Edema cerebral, contusión cerebral.
Líquido cefalorraquídeo: Hidrocefalia aguda.
Volumen sanguíneo: Hiperemia, hematomas, contusión hemorrágica.

“La presión ejercida en un fluido encerrado y en reposo se transmite uniformemente a través del volumen del fluido”

Traumatismo Craneoencefalico

DefiniciónLesión física o
deterioro funcional del
contenido craneal
Debido a un intercambio brusco
de energía mecánica

Definición
Lesión en la cabeza con: Alteración de la consciencia y/o amnesia por
cambios neurológicos o neurofisiológicos Fractura de cráneo Lesiones intracraneales Ocurrencia de muerte secundaria a la lesión en la
cabeza
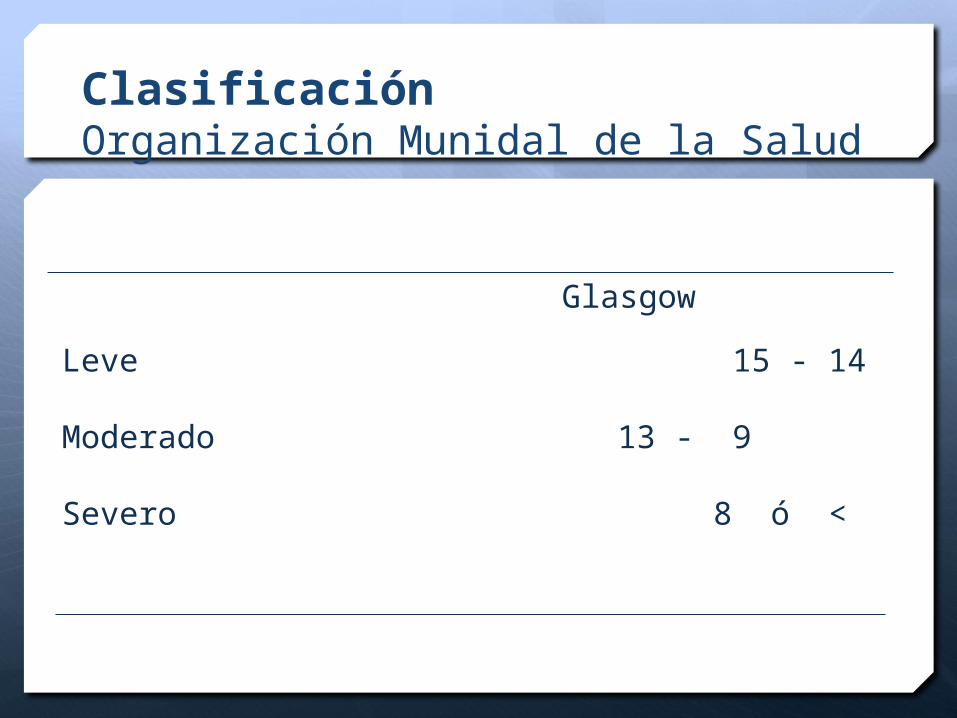
ClasificaciónOrganización Munidal de la Salud
Leve 15 - 14
Moderado 13 - 9
Severo 8 ó <
Glasgow

CO
NM
OC
ION Sacudida o
estremecimiento violento del encéfalo, con un trastorno funcional transitorio resultante
CO
NTU
SIO
N Traumatismo directo del tejido cerebral, sin solución de continuidad
CO
NC
US
IÓN Perdida
transitoria del estado de alerta sin rasgos patológicos estructurales

Fisiopatología

Fisiopatología
Injuria
Isquemia – re perfusión
Rol de caspasas y apoptosis
Respuesta inflamatoria y citocinas
Edema cerebral: vasogénico y citotóxico; acuaporinas
Coagulopatía
Hipotermia
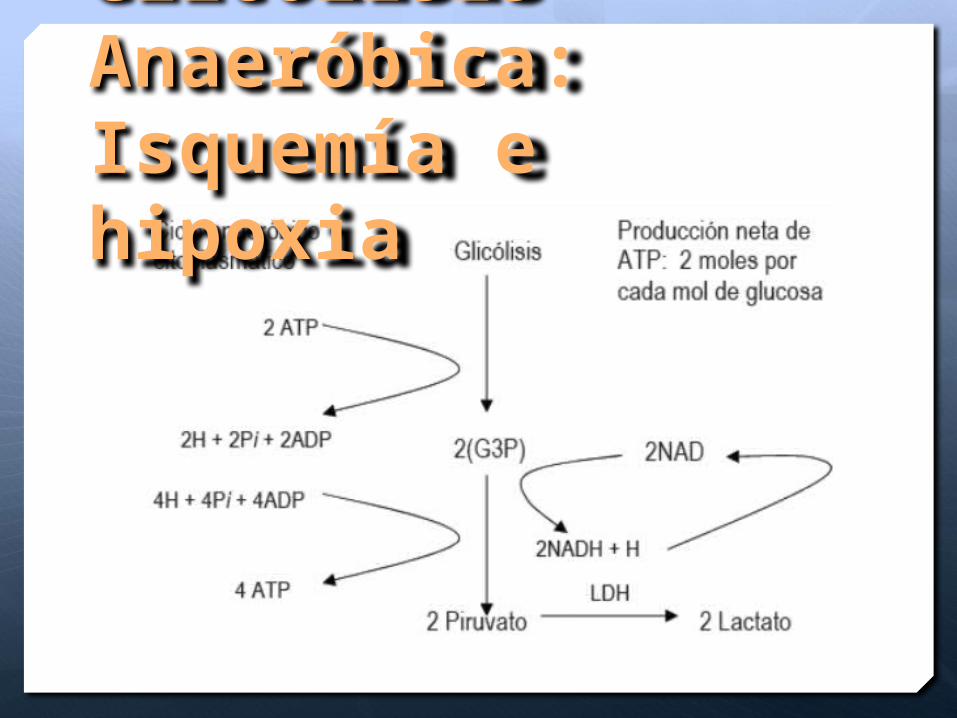
Glicolisis Anaeróbica:Isquemía e hipoxia
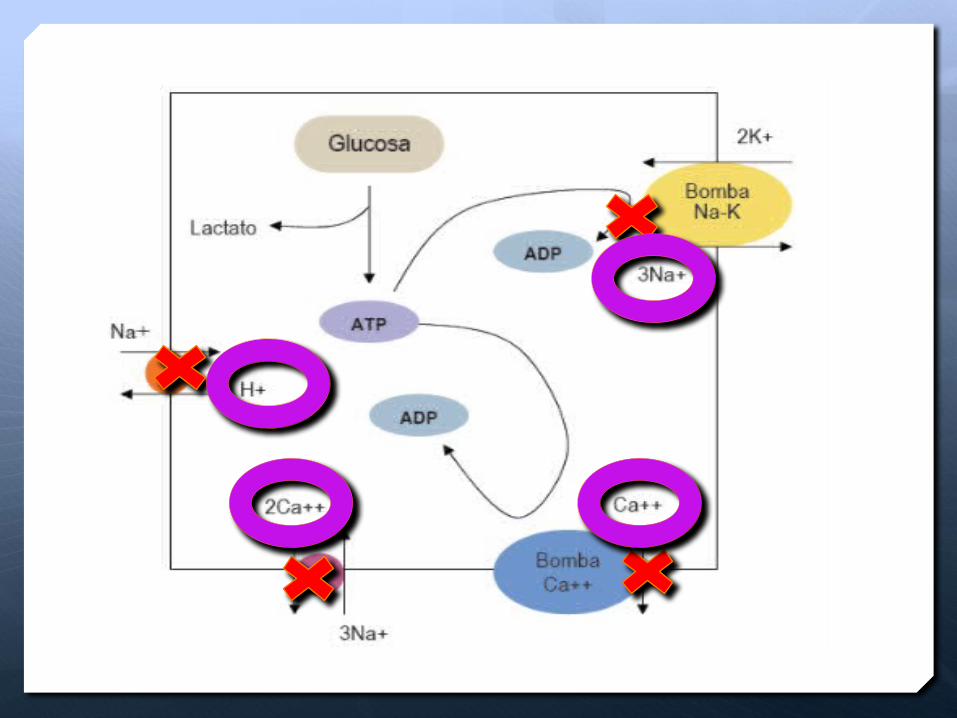
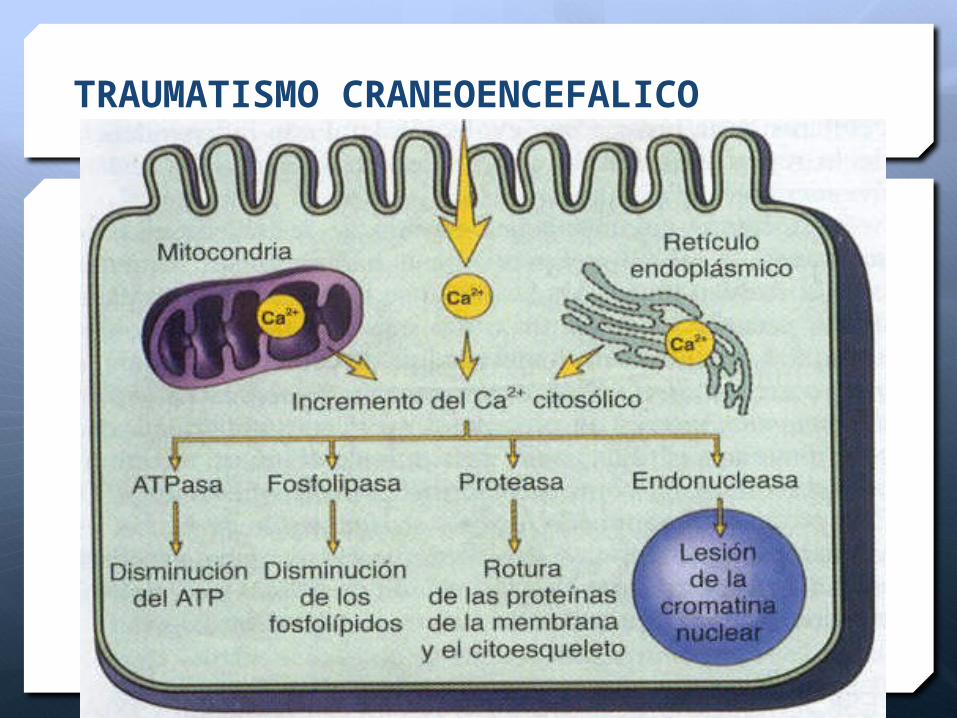
TRAUMATISMO CRANEOENCEFALICO
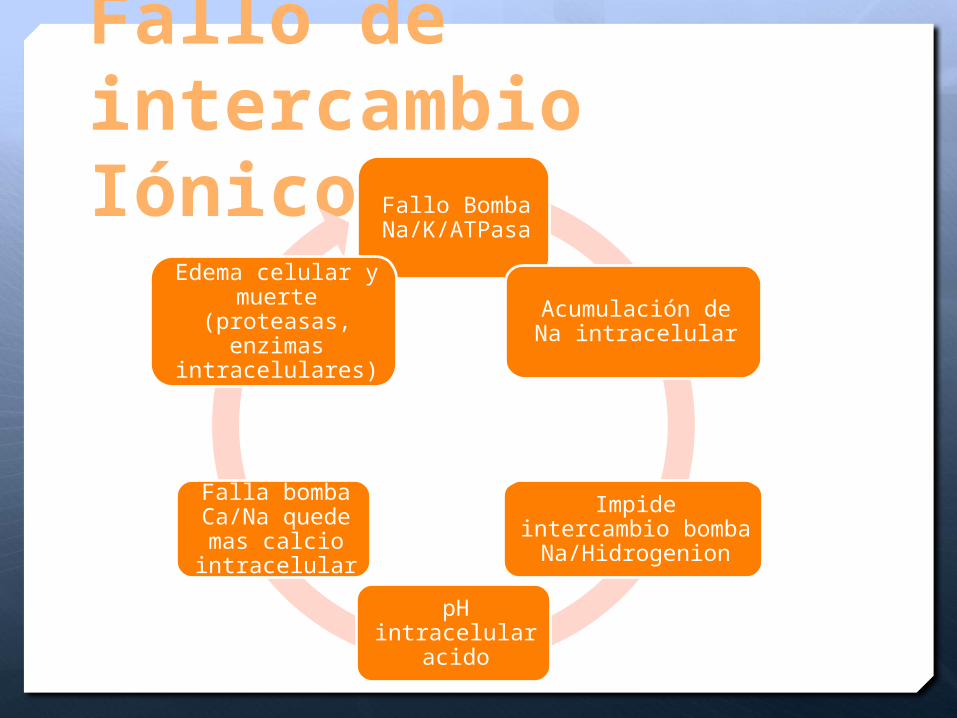
Fallo de intercambio Iónico
Fallo Bomba Na/K/ATPasa
Acumulación de Na intracelular
Impide intercambio bomba
Na/Hidrogenion
pH intracelular acido
Falla bomba Ca/Na quede mas calcio intracelular
Edema celular y muerte (proteasas,
enzimas intracelulares)
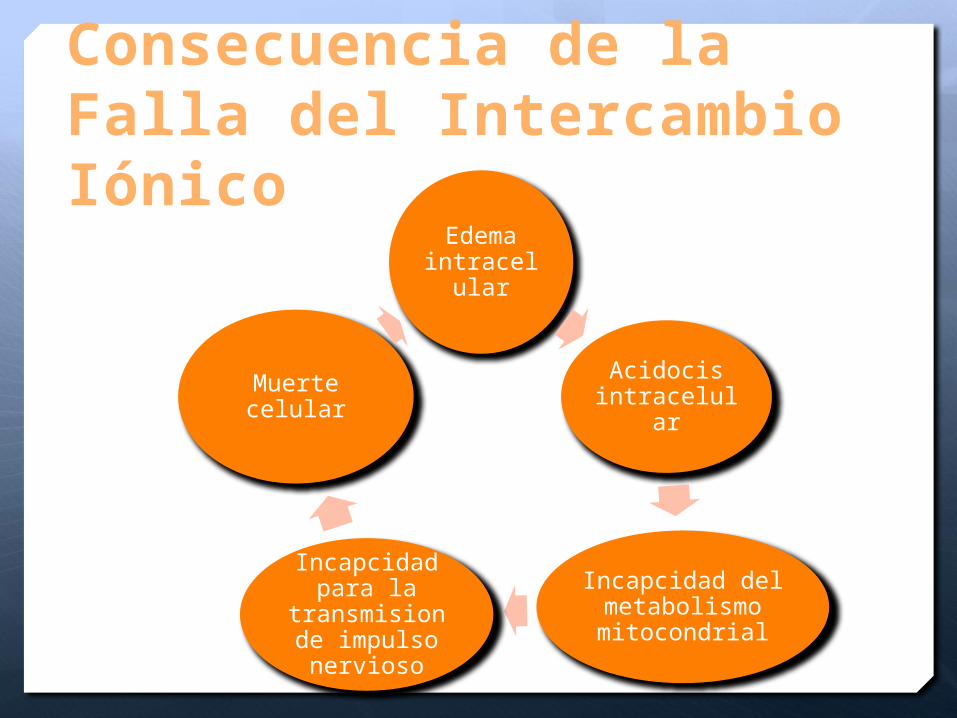
Consecuencia de la Falla del Intercambio Iónico
Edema intracelul
ar
Acidocis intracelular
Incapcidad del metabolismo mitocondrial
Incapcidad para la
transmision de impulso nervioso
Muerte celular

Disminución del ATP 3min
Depleción de los niveles por consumo en la bomba Na-K ATP asa y bomba de Calcio
Despolarización de la membrana
Aumento de Na y Ca intracelular
Edema celular
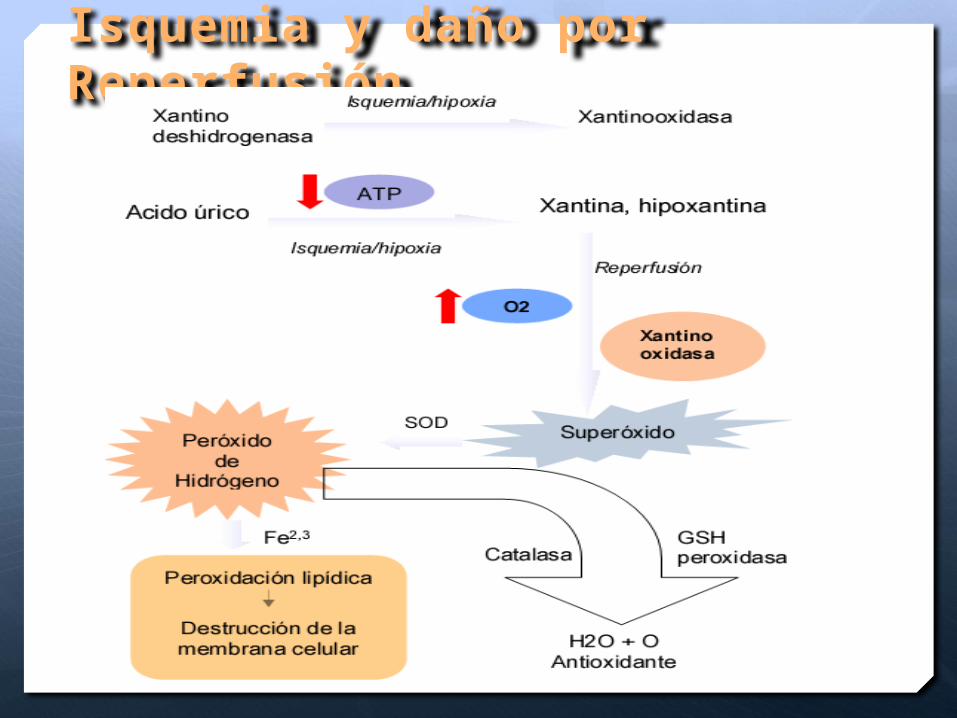
Isquemia y daño por Reperfusión
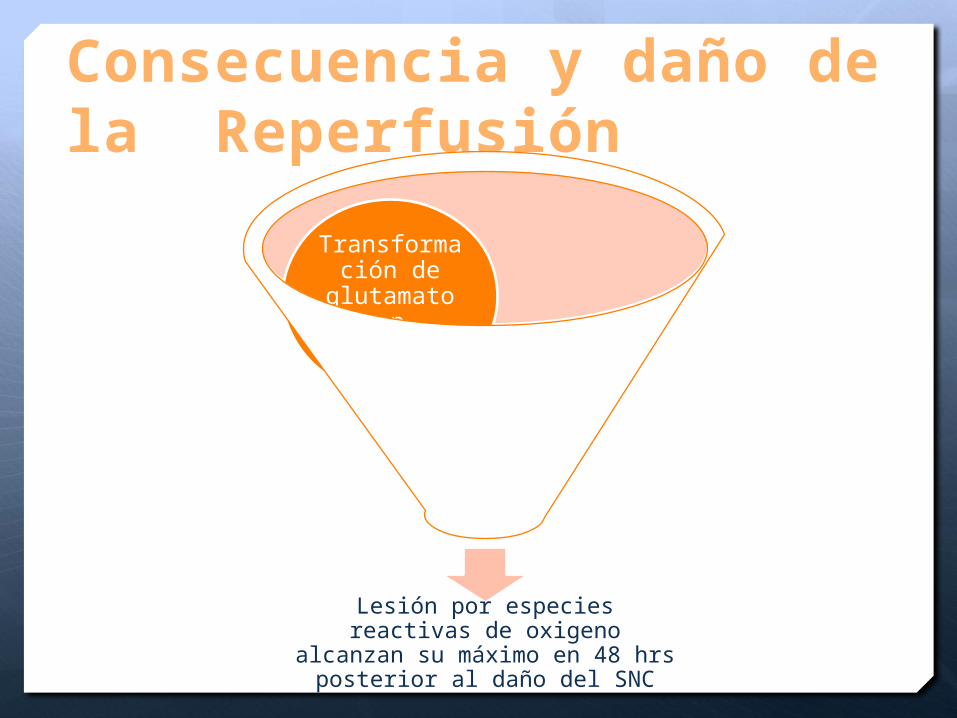
Consecuencia y daño de la Reperfusión
Lesión por especies reactivas de oxigeno alcanzan su máximo en 48 hrs posterior al daño del SNC
Destrucción de
células a través lisis
de la membrana
celular
Transformación de
glutamato en
glutamina
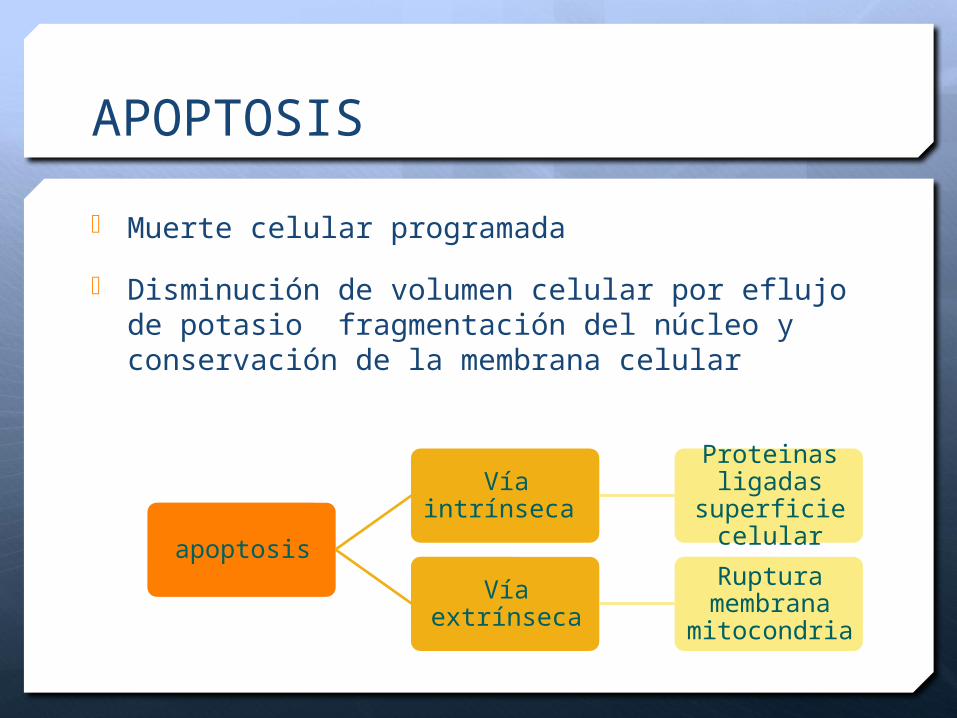
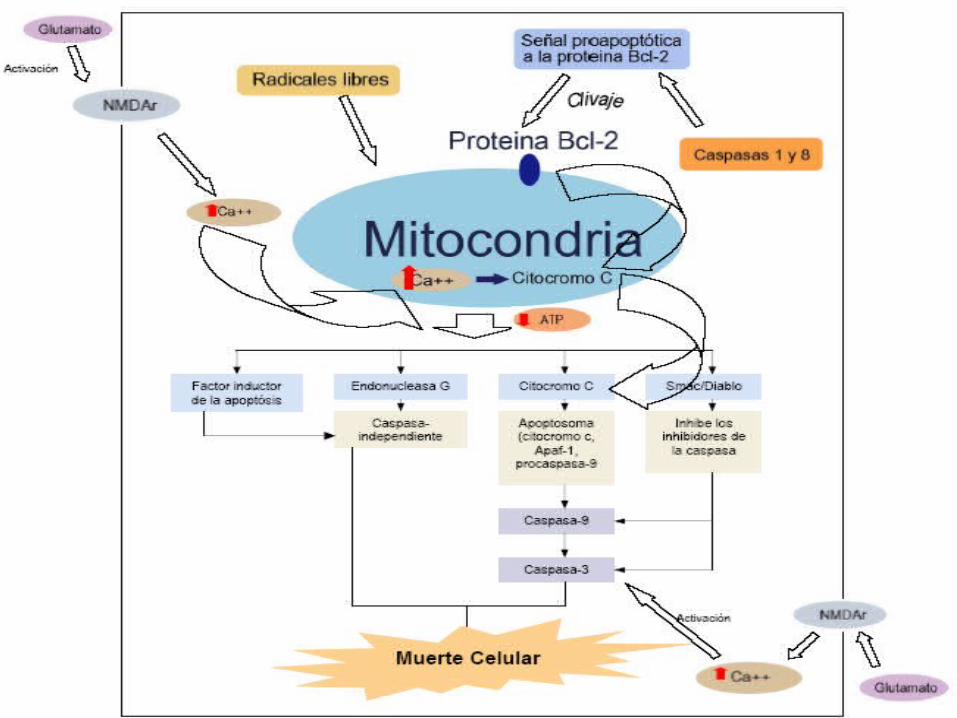
APOPTOSIS
Muerte celular programada
Disminución de volumen celular por eflujo de potasio fragmentación del núcleo y conservación de la membrana celular
apoptosis
Vía intrínseca
Proteinas ligadas
superficie celular
Vía extrínsecaRuptura
membrana mitocondria
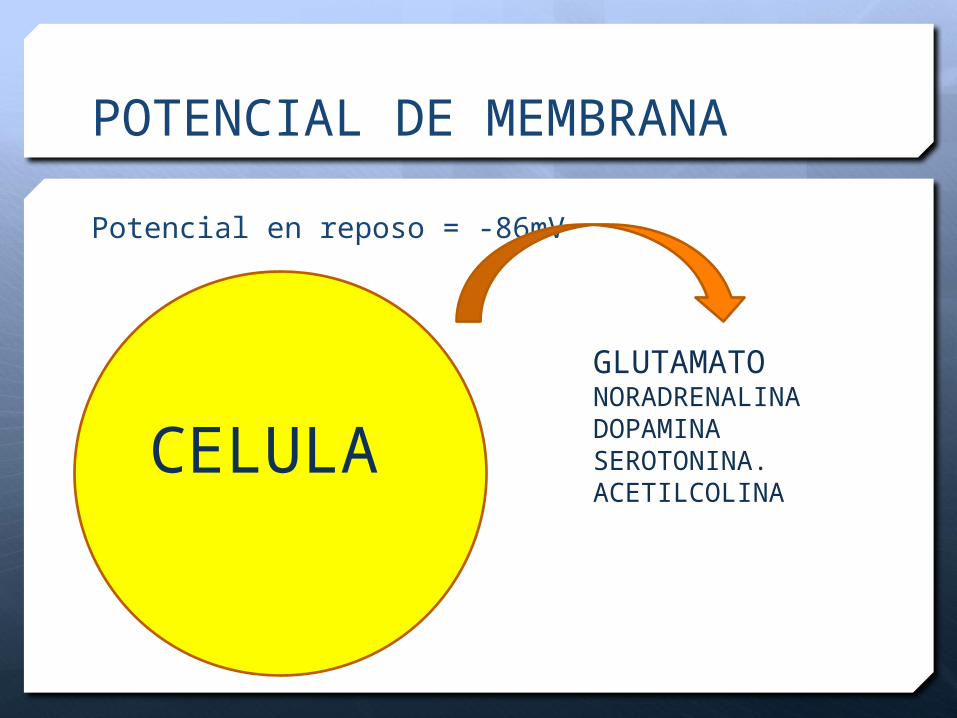
POTENCIAL DE MEMBRANA
Potencial en reposo = -86mV
GLUTAMATONORADRENALINADOPAMINASEROTONINA.ACETILCOLINA
CELULA

EXITOTOXICIDAD POR GLUTAMATO
Se activan los receptores de NMDA y producen aumento del flujo de calcio
Activación de los receptores acoplados a proteina G que liberan Ca del retículo sarcoplásmico
Activación de los receptores AMPA y aumento del flujo de sodio a la célula
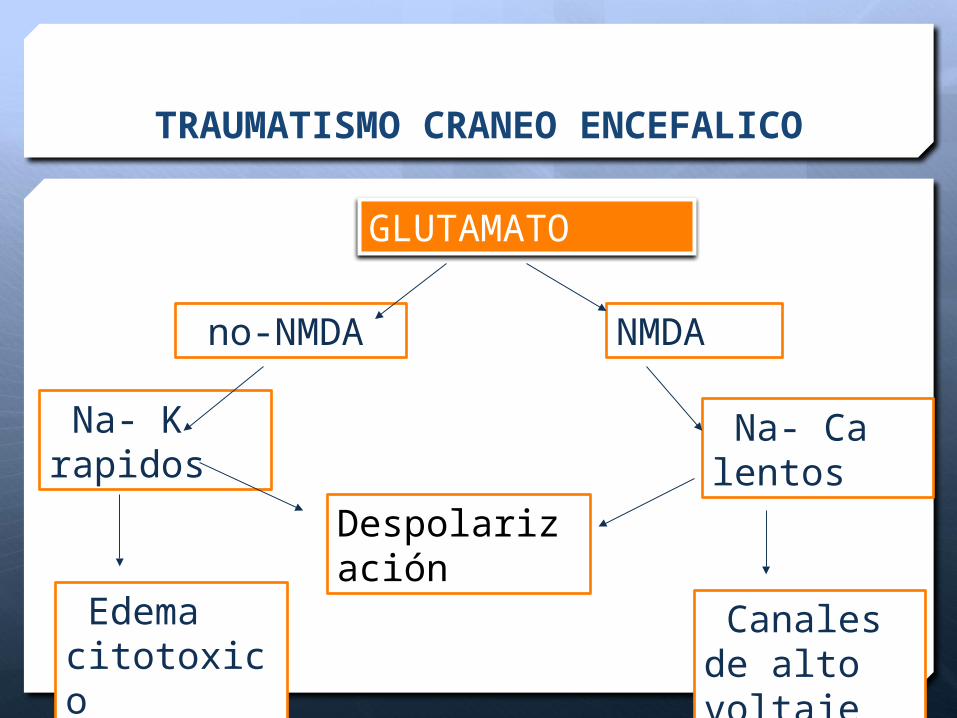
TRAUMATISMO CRANEO ENCEFALICO
GLUTAMATO
no-NMDA NMDA
Na- K rapidos
Na- Ca lentos
Edema citotoxico
Canales de alto voltaje
Despolarización

METALOPROTEASAS
Funciones reparadoras y remodeladoras sobre los componentes de la matriz extracelular (elastina, laminina, colageno, proteoglicanos)
MPM2 MPM9
Elevación a las 12-72 hrs
DISRUPCIÓN DE LA BARRERA HEMATOENCEFÁLICA
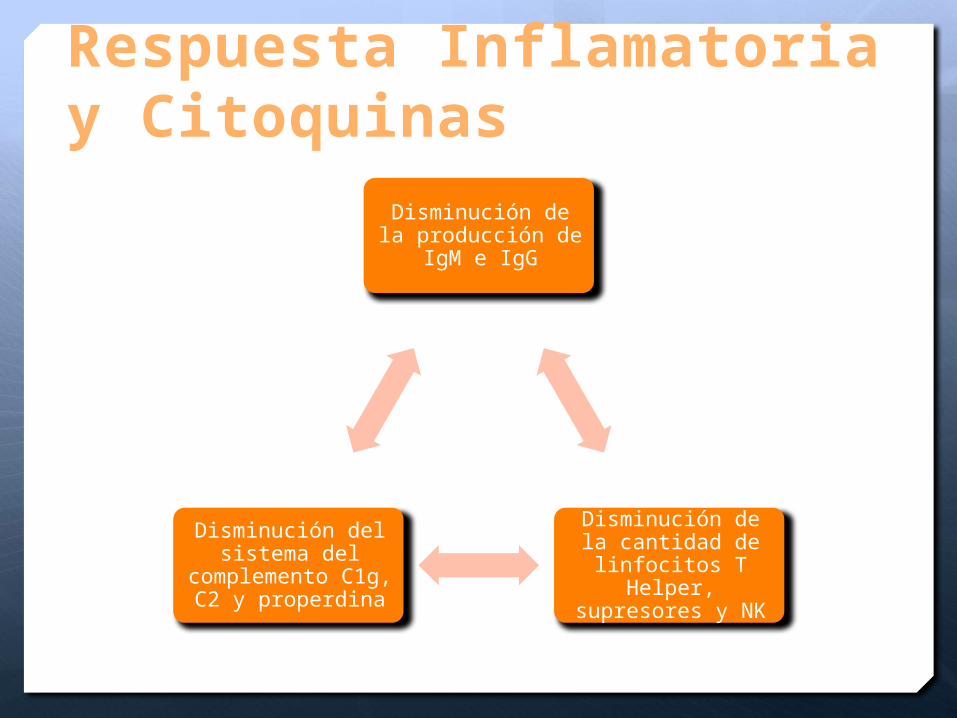
Respuesta Inflamatoria y Citoquinas
Disminución de la producción de IgM
e IgG
Disminución de la cantidad de
linfocitos T Helper, supresores y NK
Disminución del sistema del
complemento C1g, C2 y properdina
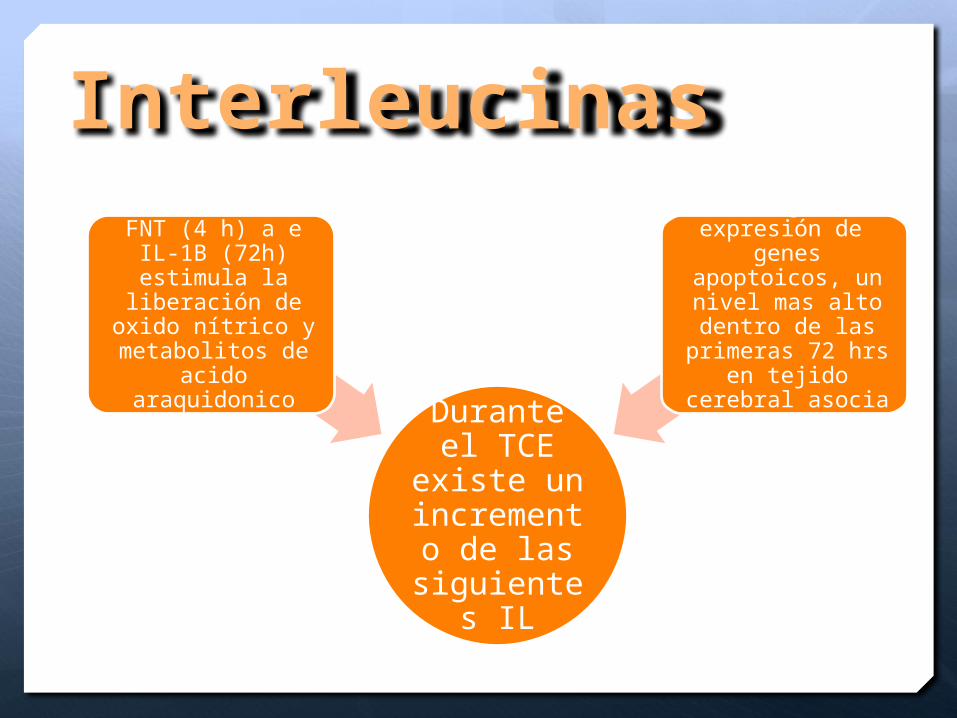
Interleucinas
Durante el TCE existe
un incremento de las
siguientes IL
FNT (4 h) a e IL-1B (72h) estimula la
liberación de oxido nítrico y
metabolitos de acido araquidonico
IL-6 Regula la expresión de
genes apoptoicos, un nivel mas alto
dentro de las primeras 72 hrs en
tejido cerebral asocia a mal pronostico

Consecuencia de la liberación de Citocinas Activación de la
apoptosis
Necrosis celular
Producción de moléculas de adhesiónDiapedesis y migración de neutrofilos
Activación y liberación de la citocinas

RESPUESTA INFLAMATORIA Y CITOCINAS
Se altera la inmunidad humoral
IgG, IgM complemento c1q, c2
75% infecciones postrauma= liberación de citocinas inflamatorias =FNTalfa, IL1, IL6.

RESPUESTA INFLAMATORIA Y CITOCINAS
Se libera acido araquidonico, PG, tromboxano, leucotrienos, ICAM 1 y selectinas,
4-72 hrs
Mal pronostico
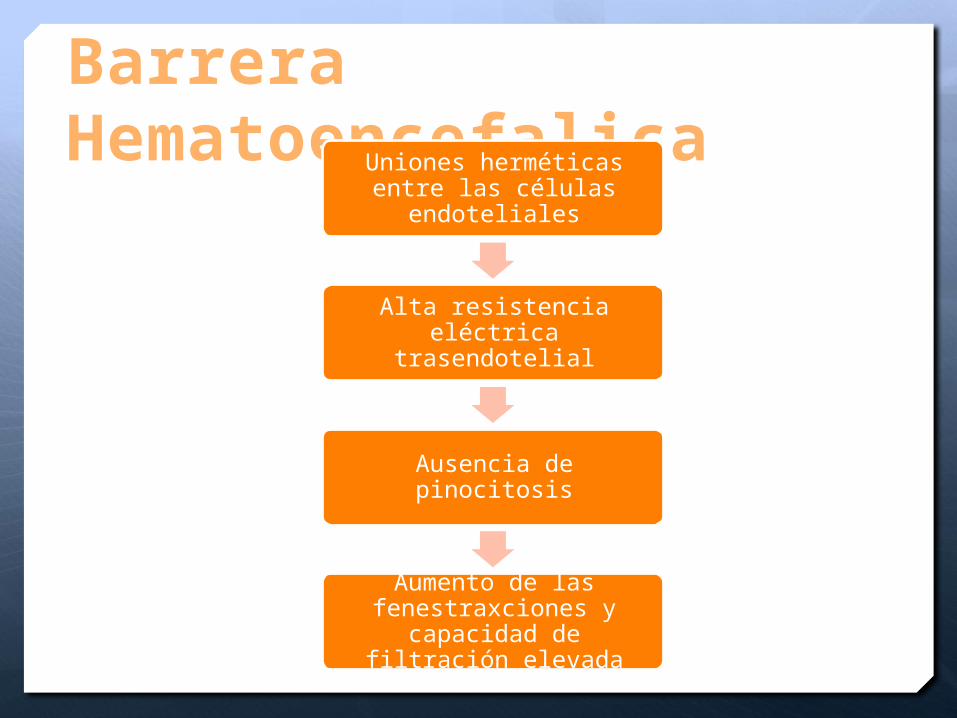
Barrera Hematoencefalica
Uniones herméticas entre las células endoteliales
Alta resistencia eléctrica trasendotelial
Ausencia de pinocitosis
Aumento de las fenestraxciones y
capacidad de filtración elevada
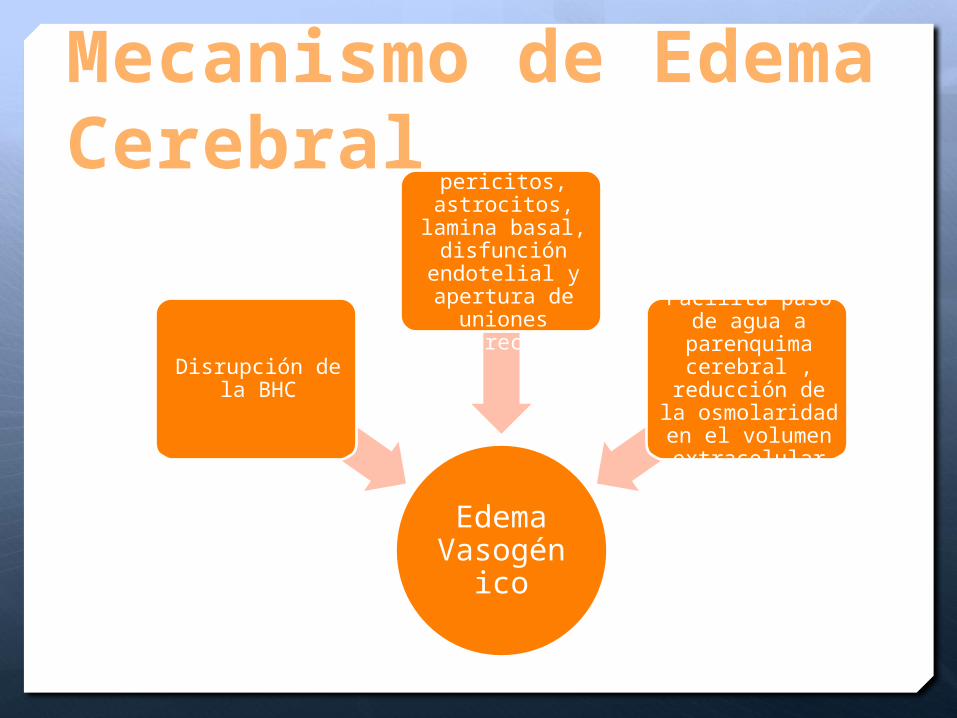
Mecanismo de Edema Cerebral
Edema Vasogéni
co
Disrupción de la BHC
Lesión de pericitos, astrocitos,
lamina basal, disfunción
endotelial y apertura de
uniones estrechas
Facilita paso de agua a
parenquima cerebral ,
reducción de la osmolaridad en
el volumen extracelular

Edema cerebral vasogénico
Disrrrupción de la barrera hematoencefálica por lesión de astrocitos, pericitos, lámina basal, lesión endotelial
Apertura de las uniones estrechas
Paso de agua → hipoosmolaridad cerebral
Edema glial perivascular
Hipoxia y edema
Predomina inicialmente en la sustancia blanca
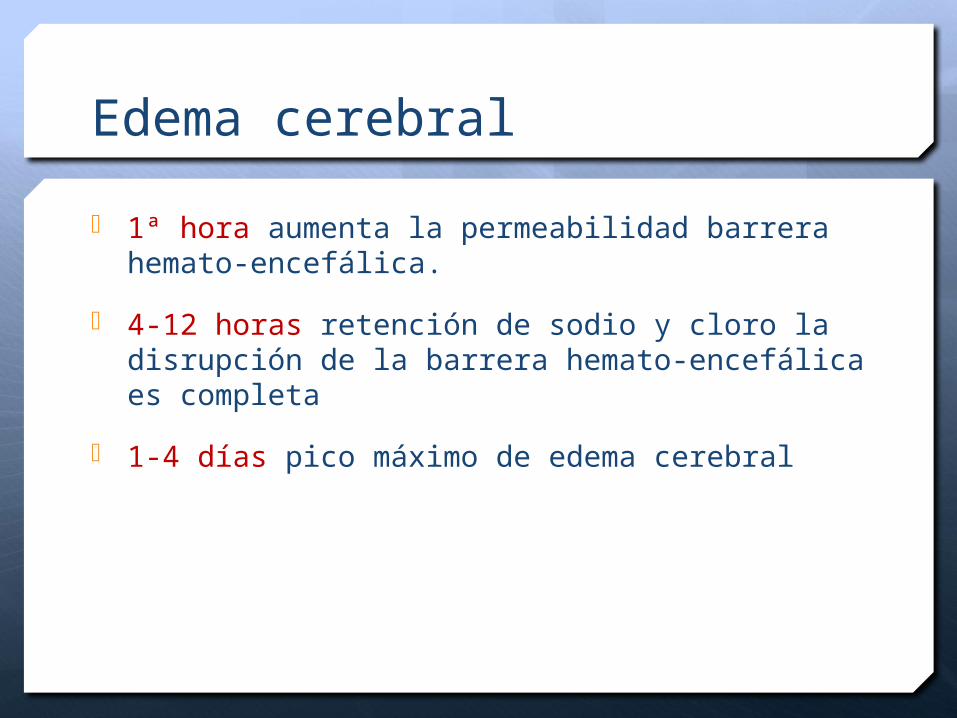
Edema cerebral
1ª hora aumenta la permeabilidad barrera hemato-encefálica.
4-12 horas retención de sodio y cloro la disrupción de la barrera hemato-encefálica es completa
1-4 días pico máximo de edema cerebral
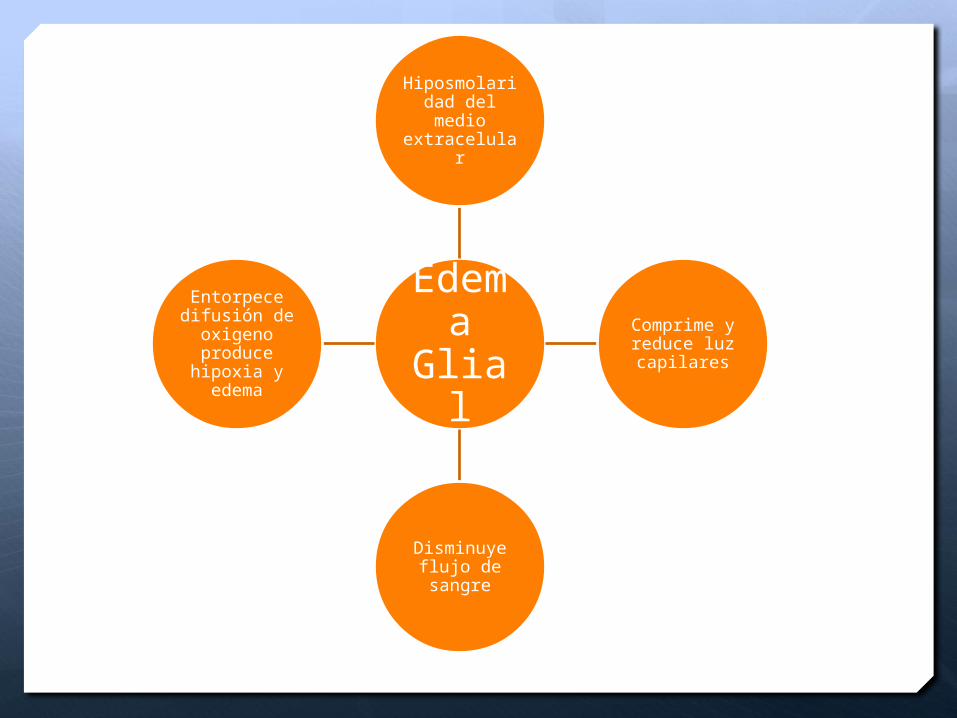
Edema
Glial
Hiposmolaridad del medio extracelular
Comprime y reduce luz capilares
Disminuye flujo de sangre
Entorpece difusión de
oxigeno produce hipoxia y edema
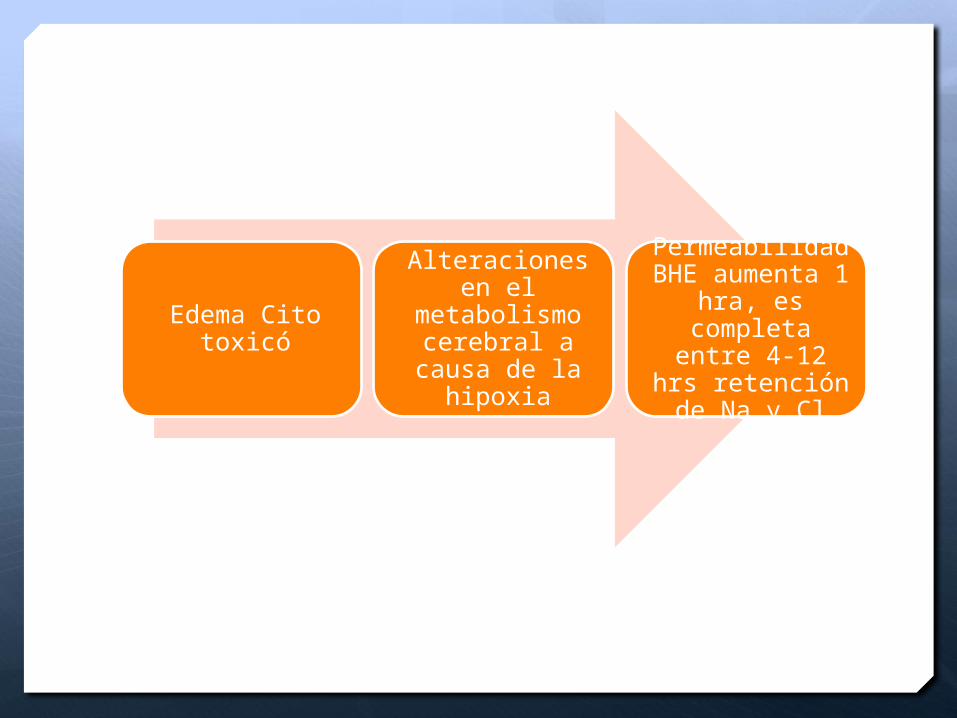
Edema Cito toxicó
Alteraciones en el metabolismo
cerebral a causa de la
hipoxia
Permeabilidad BHE aumenta 1
hra, es completa entre
4-12 hrs retención de Na
y Cl
Pico del Edema cerebral produce días 1 y 4 y posteriormente comienza a disminuir
Afecta a los compartimientos intra y extracelulares, acumulo de solutos, constituye el factor de mayor influencia en la congestión cerebral traumática
Aquaporinas
Familia de proteinas hidrofobicas
Peso molecular de 28 kDa
Modulan el paso de agua atraves de la
membrana citoplasmatica involucradas
patogenesis del edema cerebral
AstrocitosExpresan AQP4
superficie, contacto lamina basal BHE
TCE causante de la permeabilidad al
agua y generación de edema
astrocitario
Generación de edema astrocitario
TrombinaAumento de la permeabilidad
BHE
Concentración intracerebral
de Na y Cl
Alrededor de los coágulos produce una
reacción inflamatoria
Gliosis activa mediada por
trombina
Revierte al inhibir su
acción con hirudina
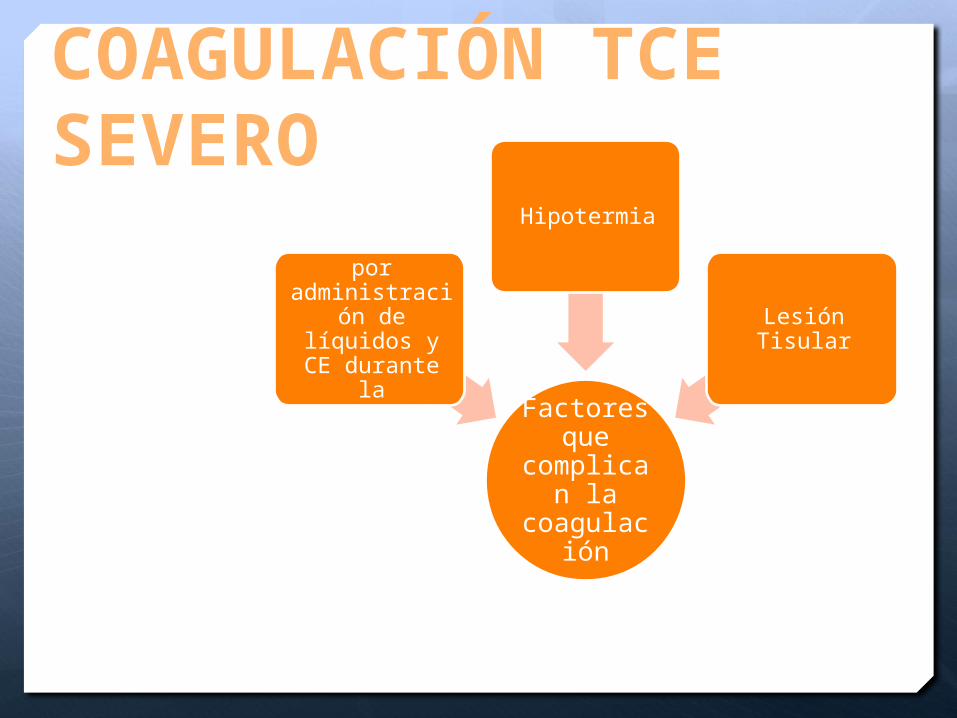
COAGULACIÓN TCE SEVERO
Factores que
complican la
coagulación
Hemodilución por
administración de líquidos y CE durante la reanimación
Hipotermia
Lesión Tisular
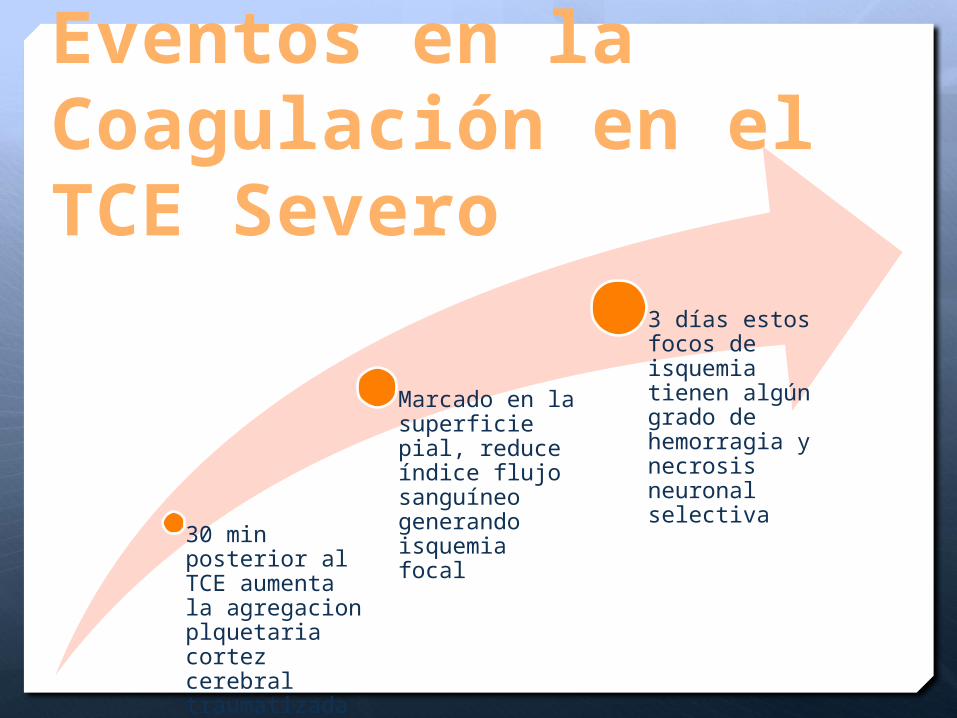
Eventos en la Coagulación en el TCE Severo
30 min posterior al TCE aumenta la agregacion plquetaria cortez cerebral traumatizada
Marcado en la superficie pial, reduce índice flujo sanguíneo generando isquemia focal
3 días estos focos de isquemia tienen algún grado de hemorragia y necrosis neuronal selectiva
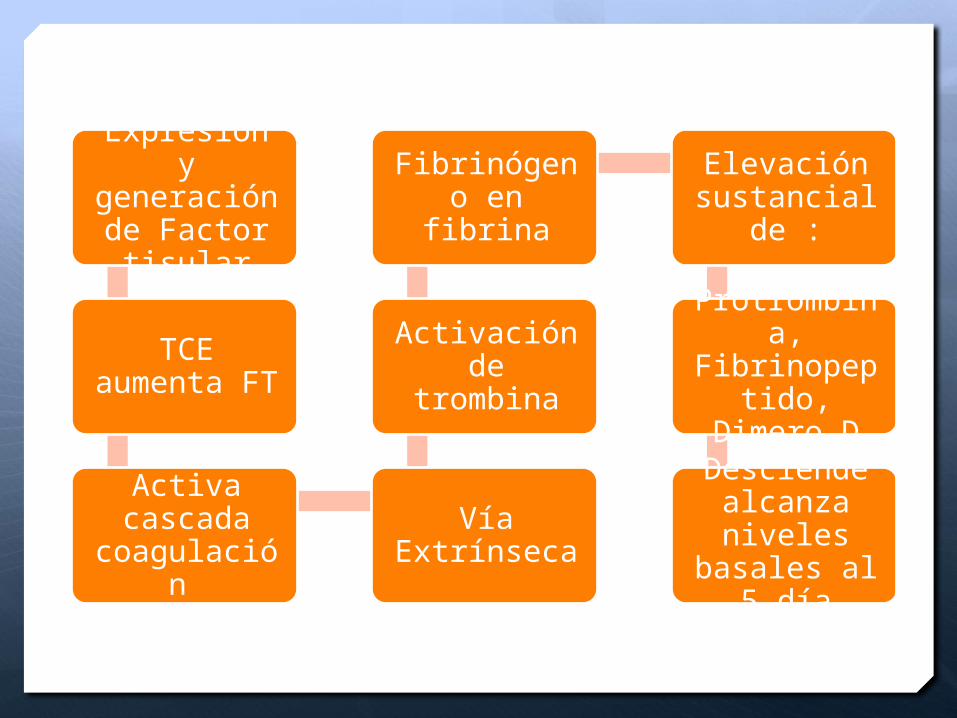
Expresión y generación de Factor
tisular
TCE aumenta FT
Activa cascada
coagulación
Vía Extrínseca
Activación de trombina
Fibrinógeno en fibrina
Elevación sustancial
de :
Protrombina, Fibrinopeptido, Dimero
D
Desciende alcanza niveles
basales al 5 día

Coagulación
30 minutos aumenta la agregación plaquetaria en la zona de corteza cerebral traumatizada.
Disminución del flujo sanguíneo y isquemia local
3 dias diferentes grados de hemorragia y necrosis neuronal selectiva
Elevación de dimero D, interleucina 6 plaquetopenia
1ª hora coagulopatía 20% trombocitopenia 14%
72 hrs coagulopatía 46% trombocitopenia 41%
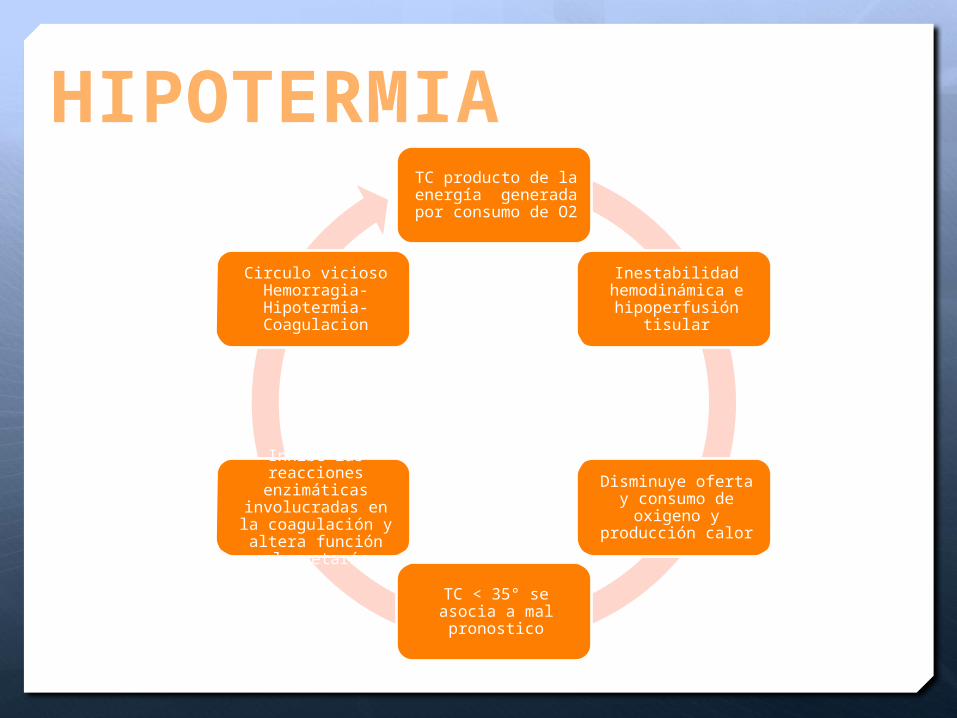
HIPOTERMIATC producto de la energía generada por consumo de O2
Inestabilidad hemodinámica e
hipoperfusión tisular
Disminuye oferta y consumo de oxigeno y producción calor
TC < 35° se asocia a mal pronostico
Inhibe las reacciones enzimáticas
involucradas en la coagulación y altera función plaquetaría
Circulo vicioso Hemorragia-Hipotermia-Coagulacion

FASES HEMODINÁMICAS
FASE I.- HIPOPERFUSIÓN; duración: 24 hrs .
FASE II: HIPEREMIA, duración: 72 hrs (del día 1 al 3).
FASE III: VASOESPASMO, duración: 10 días (del día 4 al 14).

Impacto- golpe-contragolpe, rotación
Contusión laceración cuero cabelludo
Fracturas de cráneo
Conmoción cerebral
Contusión y laceración cerebral
Hemorragia cerebral
Lesion Primaria

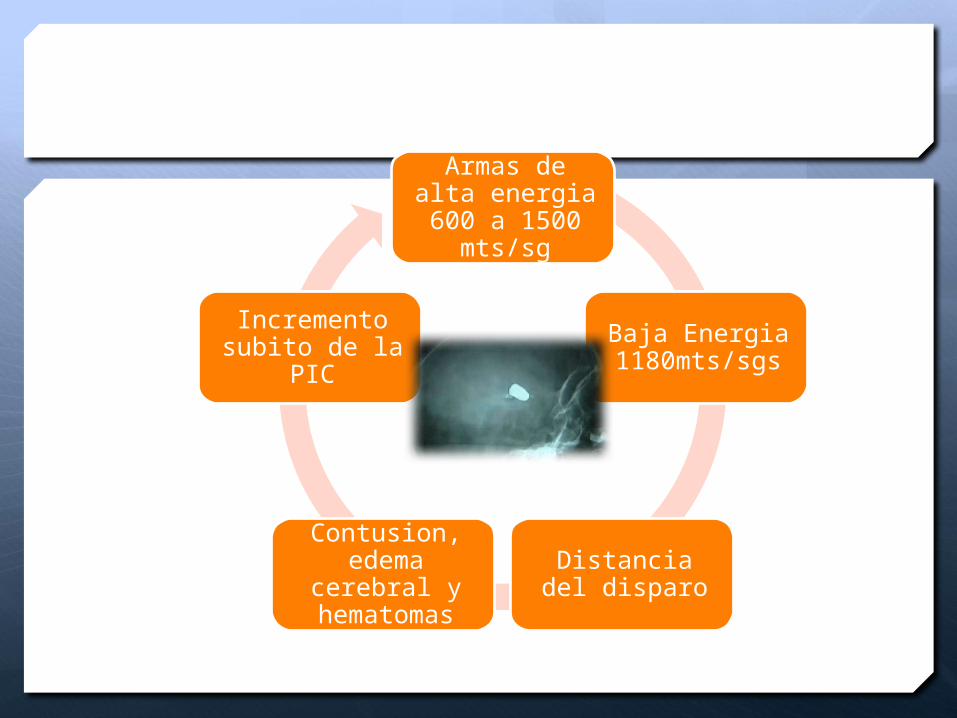
Armas de alta energia 600 a 1500 mts/sg
Baja Energia 1180mts/sgs
Distancia del disparo
Contusion, edema
cerebral y hematomas
Incremento subito de la PIC
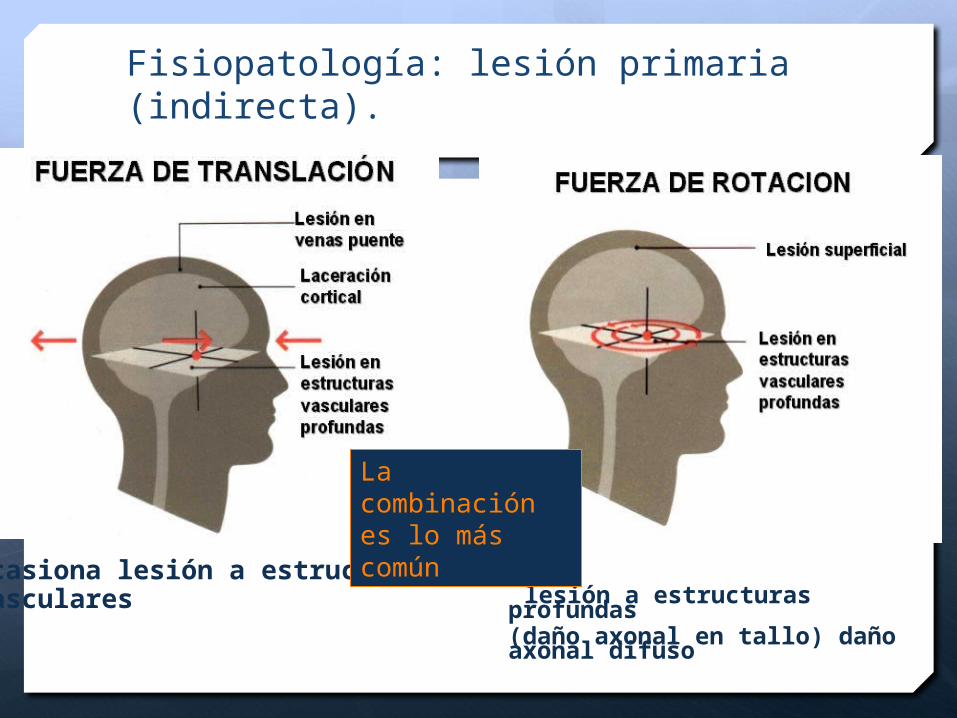
Fisiopatología: lesión primaria (indirecta).
Ocasiona lesión a estructurasvasculares lesión a estructuras profundas
(daño axonal en tallo) daño axonal difuso
La combinación es lo más común
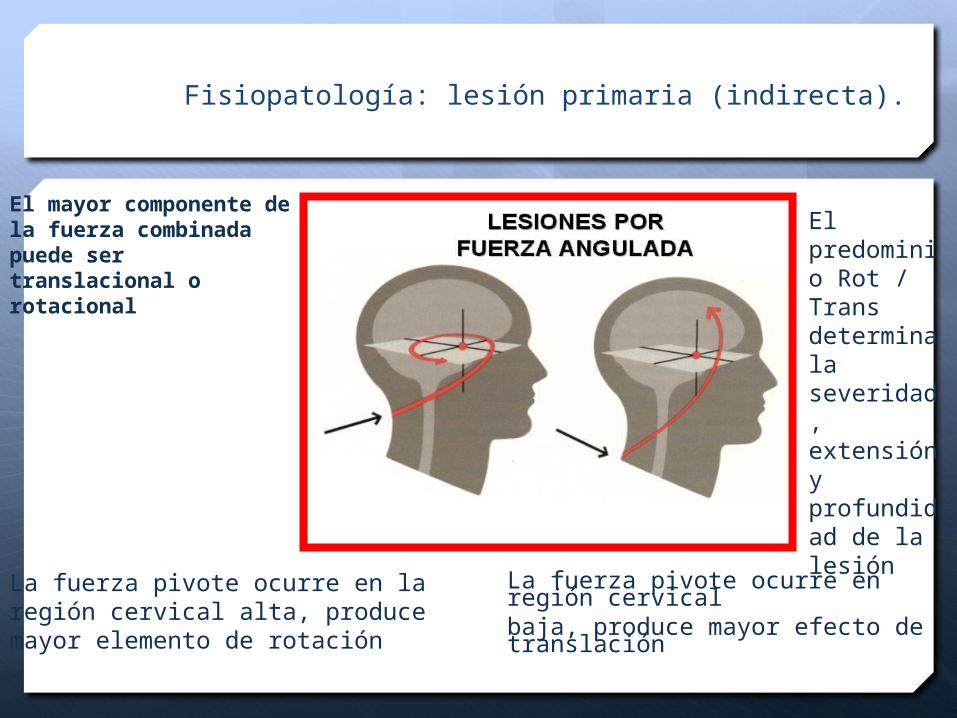
Fisiopatología: lesión primaria (indirecta).
El mayor componente de la fuerza combinada puede ser translacional o rotacional
La fuerza pivote ocurre en laregión cervical alta, producemayor elemento de rotación
La fuerza pivote ocurre en región cervicalbaja, produce mayor efecto de translación
El predominio Rot / Trans determinala severidad, extensión y profundidad de la lesión
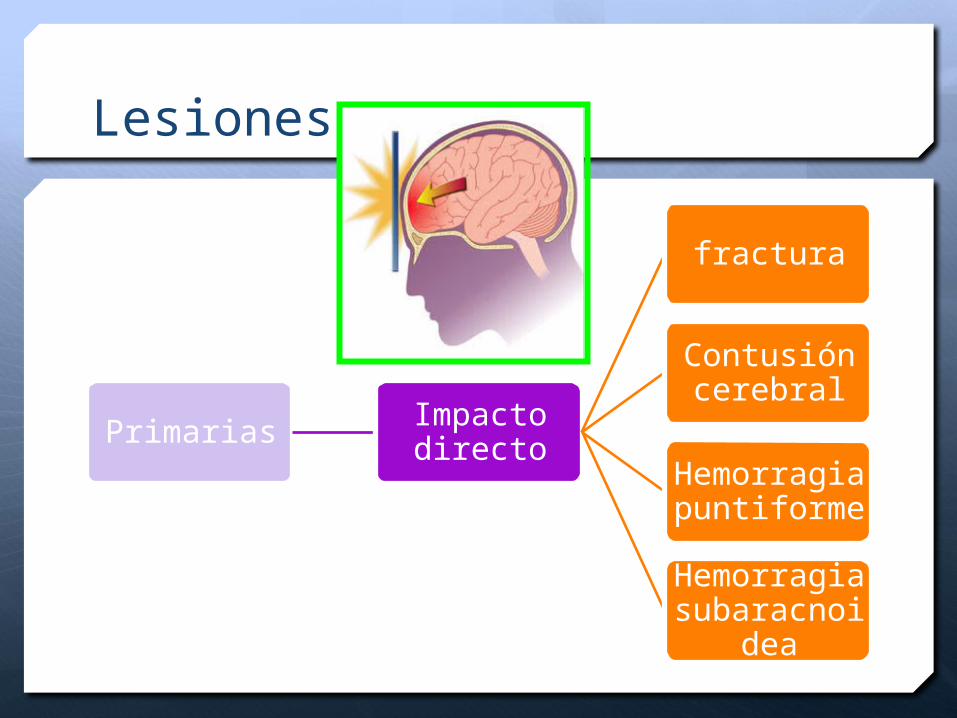
Lesiones
Primarias Impacto directo
fractura
Contusión cerebral
Hemorragia puntiforme
Hemorragia subaracnoid
ea

Lesión Secundaria

LESION CEREBRAL SECUNDARIA:
Pérdida de capacidad de regulación vasomotora cerebral – redistribución de flujo sangre, edema, isquemia.
Reducción de perfusión cerebral; aumento PIC o disminuye TAM.
Alteraciones sistémicas, hipoxemia, trastornos electrolíticos, acidosis, hipercapnia, fiebre.


















